
Синдром Тричера-Коллинза (Мандибулофациальный дизостоз, Синдром Тричера Коллинза-Франческетти, Челюстно-лицевой дизостоз)

Синдром Тричера Коллинза – это генетическая (иногда наследственная) болезнь, сопровождающаяся деформациями костей и мягких тканей лица. К симптомам относятся грубые дефекты строения лица: антимонголоидный разрез глаз, вырезки ткани век (колобомы), уменьшенные размеры челюсти и скул, гипоплазия и аномалии структур уха, расщелина или арковидная форма неба, увеличенные размеры ротовой щели и языка, слаборазвитые кости лица. Диагноз устанавливается по данным клинического осмотра, биогенетического теста и семейного анамнеза. Лечение симптоматическое, направлено на улучшение слуха, устранение жизнеугрожающих деформаций и косметических дефектов хирургическим способом.
Общие сведения
У синдрома Тричера Коллинза есть несколько синонимов: челюстно-лицевой дизостоз, синдром Тричера Коллинза-Франческетти, мандибулофациальный дизостоз. Впервые патологию описал офтальмолог из Великобритании Эдвард Тричер Коллинз в 1900 году, поэтому наиболее распространено название, соответствующее его имени. Обширный обзор заболевания был сделан в 1949 году европейскими исследователями Э. Франческетти и Д. Клейном. В настоящее время понятие «синдром Тричера Коллинза» более распространено в Великобритании и США, а термин «синдром Франческетти-Клейна» чаще используется в странах Европы. Эпидемиология болезни составляет 1:50 000. Среди мальчиков и девочек заболеваемость одинакова.

Синдром Тричера-Коллинза
Причины
Развитие синдрома в 78-93% случаев обусловлено мутациями гена TCOF1, расположенного на пятой хромосоме в регионе 5q32. Данный ген кодирует производство ядерного фосфопротеина Treacle. У 7-9% пациентов причиной заболевания является дефект гена POLR1C, локализованного на шестой хромосоме, или гена POLR1D, находящегося на тринадцатой хромосоме. Они ответственны за синтез I и III РНК-полимеразы.
При мутациях в гене TCOF1 тип наследования синдрома аутосомно-доминантный с показателем пенетрантности 90%. Это означает, что при мутации в одной хромосоме из пары вероятность проявления болезни очень высока. У больного родителя риск рождения ребенка с синдромом Тричера Коллинза составляет 50%. Возможна наследственная передача дефекта и спорадические генетические изменения (новые мутации). Экспрессивность мутации переменная – в пределах одной семьи вероятно как ослабление, так и усиление симптомов заболевания у последующих поколений. При дефектах генов POLR1C и POLR1D наследование происходит по аутосомно-рецессивному типу. В парах, где родитель имеет синдром, вероятность рождения больного малыша составляет 25%.
Патогенез
Пятая хромосома ответственна за правильное формирование скелета в период внутриутробного развития. Локализованный в ней ген TCOF1 кодирует структуру и синтез ядерного транспортного белка Treacle. Данный протеин экспрессируется в большинстве тканей организма в эмбриональном и постэмбриональном периоде, участвует в переносе генетической информации с ДНК на РНК.
В основе синдрома чаще всего лежит нонсенс-мутация, приводящая к образованию преждевременного кодона терминации и развитию гаплонедостаточности – дефицита белка, необходимого для нормального формирования лицевой части черепа. Здоровый ген обеспечивает организм белком Treacle наполовину, но такого количества недостаточно для правильного развития лицевых структур. При изменениях в генах POLR1D и POLR1C процесс транскрипции ДНК нарушается из-за недостаточности фермента-катализатора ДНК-зависимой РНК-полимеразы. Клинические проявления синдрома такие же, как и при первичной недостаточности Treacle-протеина.
Симптомы
У больных наблюдаются аномалии в строении лица. Распространенным признаком, встречающимся в 80% случаев, является двусторонняя симметричная гипоплазия скуловых костей, инфраорбитального края и нижней челюсти. Внешне это проявляется своеобразным уплощенным бесформенным лицом, на котором выделяется нос, а остальные части «утоплены» в мягких тканях. Деформация челюсти обуславливает нарушение прикуса, формирование ортогнатии (постоянно приоткрытого рта). 89% больных имеют ограниченную возможность открывания рта и антимонголоидный тип разреза глаз с заметным опущением внешнего уголка. Данные особенности частично обусловлены патологическим строением височно-нижнечелюстного сустава.
У 69% пациентов определяется колобома радужки и нижних век в промежутке между средней и внешней третью, чаще она имеет треугольную форму. Ресницы на внешнем крае нижнего века отсутствуют. Небо арковидной формы, иногда сформирована расщелина (у 28% больных). Аномалии наружного уха представлены недоразвитием или полным отсутствием ушной раковины (микротией, анотией), атрезией наружного слухового прохода и деформацией слуховых косточек. Зачастую пациенты имеют кондуктивную тугоухость. В редких случаях диагностируется энхондрома, предкозелковые фистулы, аномальное строение сердца и позвоночника.
Осложнения
Микрогнатия и стеноз верхних дыхательных путей уже в первые годы жизни могут спровоцировать проблемы при приеме пищи и трудности дыхания вплоть до удушья. Своевременная диагностика заболевания позволяет спрогнозировать эти осложнения и предпринять меры по их предупреждению. Как правило, пациенты не имеют врожденных интеллектуальных расстройств, но при отсутствии коррекции нарушений слуха становится невозможным правильное формирование речи и обучение в обычных условиях. Дети начинают отставать в умственном развитии от сверстников, имеют задержку психического развития различной степени выраженности. В связи с наличием дефектов внешности и негативным отношением окружающих больные всех возрастов относятся к группе риска по возникновению депрессии, ипохондрии, тревожности и иных невротических расстройств.
Диагностика
Диагноз может быть установлен во время беременности или сразу после рождения. Обследование показано женщинам из группы риска и детям с врожденными лицевыми деформациями. В процессе диагностики принимают участие врачи-генетики и педиатры. Синдром Тричера-Коллинза необходимо дифференцировать с другими генетическими заболеваниями, при которых существует деформация лицевой части черепа, например, с синдромом Нагера и синдромом Гольденхара. Используются следующие методы:
- Осмотр, сбор анамнеза. Определяются характерные черепно-лицевые аномалии: недоразвитость костей скул и челюсти, деформация и гипоплазия ушных раковин, антимонголоидный тип глазных щелей, нарушения слуха и дефект верхнего неба. Иногда подтвержденный диагноз синдрома имеется у одного из родителей.
- Биогенетический тест. Антенатальное исследование включает молекулярный анализ образца ворсин хориона на 10-11 неделе беременности, фетоскопию и анализ крови из сосудов плаценты на 18-20 неделе. После родов выполняется забор крови из вены ребенка. В обоих случаях исследуется ген TCOF1. Заболевание подтверждается при наличии в нем мутации любого типа.
- Дородовое УЗИ. С 20-24 недели беременности ультразвуковое исследование плода способно выявить типичные изменения лица. Наиболее четко заметна двусторонняя аномалия ушей, гипоплазия скул и челюсти.
Дополнительно назначаются обследования, позволяющие своевременно обнаружить жизнеугрожающие состояния, оценить степень деформации костей черепа. Определяется эффективность кормления ребенка, уровень насыщения гемоглобина кислородом, ритмичность и глубина дыхания. Для диагностики сохранности слуха на 5-6 день жизни проводится аудиологическое тестирование. Инструментальная диагностика включает рентгенографию черепа, КТ и МРТ головного мозга.
Лечение синдрома
Специфической терапии не существует. Лечение нацелено на устранение симптомов и последствий заболевания, предполагает проведение хирургических операций и реабилитационных мероприятий. Объем процедур и сроки их выполнения устанавливаются индивидуально с учетом наличия угрозы для жизни больного, противопоказаний и рисков, связанных с оперативным вмешательством. Общая схема лечения включает:
- Восстановление глотания и дыхания. При развитии респираторного дистресс-синдрома осуществляется трахеостомия, дистракция подвижной челюсти, неинвазивная вентиляция легких. При невозможности потребления пищи устанавливается гастростома.
- Восстановление слуха. Деформация наружного и среднего уха устраняется хирургическим путем, но потеря слуха чаще обусловлена повреждением слуховых мелких косточек, поэтому оперативные вмешательства с целью устранения тугоухости неэффективны. Предпочтительна реабилитация слуховыми аппаратами.
- Устранение внешних дефектов. Деформации корректируются методами пластической и нижнечелюстно-лицевой хирургии. Применяется липоскульптурирование, хирургическая дистракция костей, установка трансплантатов и хирургическое восстановление неба.
Прогноз и профилактика
Комплексное лечение и реабилитация значительно улучшают качество жизни больных. При легкой и умеренной выраженности синдрома прогноз благоприятный. Профилактика затруднена, поскольку заболевание является генетическим, а мутации способны возникать спонтанно. Супружеским парам, в которых один родитель болен, необходимо медико-генетическое консультирование и перинатальная диагностика синдрома на ранних сроках беременности. Для снижения риска вынашивания больного ребенка рекомендуется процедура экстракорпорального оплодотворения с предварительным отбором генетически здоровых эмбрионов.
|
Литература 1. Медицинская и клиническая генетика для стоматологов: учебник для вузов. Под ред. О. О. Янушевича – 2009. 2. Особенности стоматологической патологии при некоторых наследственных заболеваниях / Шишкова О.В., Максимова Ю.В. // Медицина и образование в Сибири – 2007 — №3. |
Код МКБ-10 Q75.4 |
Синдром Тричера-Коллинза — лечение в Москве
Синдром Тричера Коллинза, также известный как челюстно-лицевой дизостоз, является редким генетическим заболеванием, вызванным мутацией в пятой хромосоме
Он никак не влияет на умственное развитие ребенка, характеризуется частичной атрофией скул, деформацией нижней челюсти, раздвоением век и запавшими косыми глазами. Из-за искаженных ушных раковин у пациентов часто возникают проблемы со слухом или полная глухота.
Проблемы людей с синдромом Тричера Коллинза
Помимо очевидных физических недостатков, болезнь приносит эмоциональные проблемы – люди с синдромом Тричера Коллинза часто имеют низкую самооценку и страдают неуверенностью в себе. Их нетипичный внешний вид зачастую вызывает неоднозначное отношение окружающих и может привести к социальной изоляции, что особенно тяжело для детей и подростков. «Люди без лица» обычно не любят смотреть в зеркало и могут чувствовать себя чужими в мире, где нет никого, похожего на них.
О том, как живут люди с синдромом Тричера Коллинза, можно узнать, посмотрев мелодраму Стивена Чбоски «Чудо-мальчик» – экранизацию самого продаваемого романа американской писательницы Р. Дж. Паласио, разошедшегося миллионными тиражами по всему миру. Его главный герой Огги Пуллман – пятиклассник с синдромом Тричера-Коллинза, который не расстается со своим игрушечным шлемом астронавта, позволяющим избегать любопытных, и не всегда доброжелательных, взглядов прохожих. За первые 10 лет жизни мальчик перенес 27 операций, позволивших ему дышать, видеть и слышать.
Что такое синдром Тричера-Коллинза?
Синдром Тричера-Коллинза (сокр. СТК, челюстно-лицевой дизостоз) — редкое генетическое заболевание, характеризующееся отличительными аномалиями головы и лица. Черепно-лицевые аномалии имеют тенденцию вызывать недоразвитие скулового комплекса, скул, челюстей, неба и рта, что может привести к затруднению дыхания и кормлению. Кроме того, у больных могут быть пороки развития глаз, включая наклоненные вниз отверстия между верхними и нижними веками (глазные щели) и аномалии структур наружного и среднего уха, которые могут привести к потере слуха.
Мозг и поведенческие аномалии, такие как микроцефалия и психомоторная задержка, также иногда наблюдаются при синдроме. Конкретные симптомы и физические признаки, связанные с СТК, могут сильно различаться. Некоторые люди могут быть настолько слабо затронуты болезнью, что их состояние может остаться не диагностированным, в то время как у других могут развиться серьезные, угрожающие жизни осложнения.
Хотя СТК в первую очередь возникает в результате изменения (мутации) в гене TCOF1, она также связана с мутациями в генах POLR1B, POLR1C или POLR1D. В случае TCOF1 и POLR1B тип наследования является аутосомно-доминантным (вид наследования, при котором генетическая обусловленная болезнь проявляется в случае, если у человека есть хотя бы один соответствующий ей «дефектный» ген, причем этот ген не содержится в половых (Х и Y) хромосомах), в то время как POLR1C наследуется по аутосомно-рецессивному типу (вид наследования, при котором генетически обусловленная болезнь проявляется в том и только в том случае, если «дефектный» ген был унаследован от обоих родителей и при этом не содержится в половых (Х и Y) хромосомах). Напротив, мутация в гене POLR1D, может быть как аутосомно-доминантная, так и аутосомно-рецессивная.
Синдром назван в честь Эдварда-Тричера-Коллинза, лондонского офтальмолога, который впервые описал это расстройство в медицинской литературе в 1900 году. СТК также известен как челюстно-лицевой дизостоз или синдром Тричера-Коллинза-Франческетти.
Признаки и симптомы
Симптомы и тяжесть синдрома Тричера-Коллинза могут значительно варьироваться от одного человека к другому, даже среди членов одной семьи. У некоторых людей симптомы могут быть слабовыражены, другие могут иметь значительные нарушения и потенциально опасные для жизни респираторные осложнения. Важно отметить, что у пострадавших людей не будет всех симптомов, обсуждаемых ниже.
Основные характерные черты СТК охватывают определенные кости лица, ушей и мягких тканей вокруг глаз. Пострадавшие люди имеют отличительные черты лица и потенциально могут иметь проблемы со слухом и зрением. Нарушения при СТК, как правило, симметричны (почти идентичны с обеих сторон лица) и присутствуют при рождении (врожденные). Развитие речи и языка может быть нарушено потерей слуха, расщелиной неба или челюсти и проблемами с дыхательными путями. На интеллект они обычно не влияют, но аномалии мозга и поведения, такие как микроцефалия и когнитивная задержка наблюдаются часто.
У детей с синдромом наблюдаются недоразвитость или отсутствие скул, в результате чего эта область лица выглядит плоской или впалой. Кость нижней челюсти развивается не полностью (гипоплазия нижней челюсти), в результате чего подбородок и нижняя челюсть выглядят аномально маленькими (микрогнатия). Некоторые костные структуры (например, венечные и кондилоидные отростки), которые соединяют участки кости нижней челюсти к мышце, могут быть необычно плоскими или отсутствовать. У пострадавших детей может также наблюдаться недоразвитость горла (гипоплазия глотки). Гипоплазия глотки с недоразвитостью нижней челюсти (гипоплазия нижней челюсти) и/или аномальная малость челюсти (микрогнатия) может способствовать проблемам с питанием и/или затруднению дыхания (дыхательная недостаточность) в раннем детстве. Дети могут испытывать обструктивное апноэ во сне, для которого характерны повторяющиеся кратковременные нарушения нормального дыхания и движения воздуха во время сна. У некоторых больных с серьезными поражениями могут возникнуть опасные для жизни проблемы с дыханием.

Дополнительные нарушения, которые могут способствовать затруднению дыхания или кормлению, включают сужение или обструкцию носовых дыхательных путей. Некоторые дети могут иметь признаки «последовательности Пьера Робина», которые включают тяжелую микрогнатию, язык, который смещен дальше назад во рту, чем обычно (глоссоптоз), с неполным закрытием крыши рта (волчья пасть) или без него. Даже у пациентов, где нёбо срастается, оно может оставаться выпуклым, что может повлиять на питание и дыхание. Кроме того, пороки развития рта и челюсти могут привести к зубным аномалиям, таким как неправильное прикус. Также сообщалось о дополнительных зубных аномалиях, включая отсутствие зубов.
У людей с СТК может развиться потеря слуха из-за неспособности звуковых волн проходить через среднее ухо (кондуктивная потеря слуха). Кондуктивная потеря слуха обычно возникает из-за аномалий, влияющих на структуры в среднем ухе, и у людей с СТК также могут быть неправильно сформированные или отсутствующие косточки, три маленьких кости, через которые звуковые волны передаются в среднее ухо. Кроме того, наружные ушные структуры часто отсутствуют. Внешние уши могут быть смяты или повернуты. Напротив, внутреннее ухо обычно не поражено, хотя сообщалось о мальформации костного спирального органа во внутреннем ухе (улитки) и структур во внутреннем ухе, которые играют роль в равновесии (вестибулярный аппарат). Дополнительные симптомы могут включать присутствие небольших наростов кожи или ямок непосредственно перед наружным ухом (доаурикулярные метки) и ненормального прохода, который закрыт на одном конце (слепой свищ), который обычно отводит уши к носу.
Многие дети с СТК имеют аномалии тканей, окружающих глаза. Эти различия в глазах могут дать пострадавшим пациентам печальный внешний вид лица. Наиболее распространенным глазным симптомом является наклон вниз к отверстию между верхним и нижним веками (глазные щели). Дополнительные симптомы включают в себя выемку нижнего века или расщелину отсутствующей ткани век (колобома век), частичное отсутствие ресниц на нижнем веке, косоглазие и суженные слезные протоки (дакростеноз). Иногда наблюдаются пороки развития глазного яблока, которые могут включать надрез или расщелину отсутствующей ткани радужной оболочки или аномально маленьких глаз (микрофтальмия). У некоторых пациентов может возникнуть потеря зрения. Степень нарушения зрения варьируется в зависимости от тяжести и сочетания глазных аномалий. Нарушение нижнего века может привести к высыханию глаз, что увеличивает риск хронического раздражения и глазных инфекций.
Приблизительно у 5% людей с СТК наблюдается дефицит развития или неврологические проблемы, такие как психомоторная задержка. Однако интеллект, как правило, не влияет на нормальное развитие языка. Тем не менее, проблемы с развитием речи могут возникать из-за потери слуха, расщепления нёба или трудностей с воспроизведением звуков из-за структурных искажений. У некоторых больных с СТК наблюдаются дополнительные физические отклонения, такие как широко расставленные глаза, насечка верхнего века, деформация носа, ненормально широкий рот (макростомия), необычный рост волос на голове в направлении щек, врожденные пороки сердца и/или желудочно-кишечные пороки развития.
Причины
СТК вызывается мутацией генов TCOF1, POLR1B, POLR1C или POLR1D. В случае мутации гена TCOF1 тип наследования является аутосомно-доминантным, хотя наблюдались очень редкие случаи аутосомно-рецессивных мутаций. Мутации POLR1B являются аутосомно-доминантными, тогда как при POLR1C они являются аутосомно-рецессивными, а мутации POLR1D могут быть аутосомно-доминантными или аутосомно-рецессивными.
Генетические заболевания определяются сочетанием генов для определенного признака, которые находятся на хромосомах, полученных от отца и матери. Доминантные генетические нарушения возникают, когда для появления заболевания необходима только одна копия ненормального гена. Для TCOF1, POLR1B и POLR1D аномальный ген может быть унаследован от любого из родителей или может быть результатом новой мутации (спонтанного изменения гена) у пострадавшего человека. Примерно у 60% пациентов с синдромом Тричера-Коллинза мутация является новой, которая возникает случайно (спонтанно) без предшествующего семейного анамнеза заболевания (мутация de novo). Тем не менее, родители также могут быть слегка затронуты и не знать, что у них есть расстройство. Риск передачи ненормального гена от пострадавшего родителя к потомству составляет 50% при каждой беременности. Риск одинаков для детей мужского и женского пола. Независимо от того, наследуется ли мутация от матери или отца, похоже, что она не имеет отношения к тяжести состояния СТК у их детей.
Рецессивные генетические нарушения (например, СТК, вызванные мутациями POLR1C или POLR1D) возникают, когда человек наследует один и тот же аномальный ген для одного и того же признака от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, человек будет носителем заболевания, но обычно не проявлять симптомов. Риск для двух родителей-носителей, чтобы передать оба дефектных гена и, следовательно, родить больного ребенка, составляет 25% при каждой беременности. Риск зачать ребенка, который будет носителем, как родители, составляет 50% при каждой беременности. Вероятность для ребенка получить нормальные гены от обоих родителей и быть генетически незатронутым для этой конкретной болезни составляет 25%.
Мутации гена TCOF1 вызывают большинство (приблизительно 80%) случаев синдрома Тричера-Коллинза. TCOF1 содержит инструкции, которые кодируют (создают) белок, известный как treacle. Точная роль, которую играет белок treacle в развитии СТК, полностью не понята. Исследователи определили, что treacle играет роль в создании определенных небольших структур в клетках, которые собирают белки (рибосомы). Это особенно важно для формирования группы клеток, называемых клетками нервного гребня, которые образуются очень рано во время эмбрионального развития и дают начало большей части кости и хряща, лежащих под лицом.
Состояния, которые возникают из-за дефектов в образовании (биогенезе) рибосом, называются рибосомопатиями. POLR1B кодирует субъединицу РНК-полимеразы 1, тогда как POLR1C и POLR1D кодируют субъединицы РНК-полимеразы I и III, каждая из которых также важна для биогенеза рибосом. Вероятно, что мутации в TCOF1, POLR1B, POLR1C и POLR1D вызывают недостаточную сборку белка и не позволяют определенным клеткам нервной системы и нервного гребня удовлетворять свои потребности в пролиферации и росте во время развития эмбриона. Поскольку СТК сильно различается, исследователи предполагают, что дополнительные генетические и, возможно, экологические факторы также могут играть роль в варьируемой степени тяжести заболевания. В поддержку этой концепции недавние экспериментальные данные показали, что белок treacle играет критическую роль в защите от вызванного окислительным стрессом повреждения ДНК в нервных клетках, а также в ориентации гребня во время деления нервных клеток, которые впоследствии влияют на развитие головы и лица.
Затронутые группы населения
Синдром Тричера-Коллинза затрагивает как мужчин, так и женщин в равных количествах. По оценкам, распространенность составляет от 1 на 10 000-50 000 человек в общей популяции. Некоторые люди с легкими проявлениями болезни могут не диагностироваться, что затрудняет определение истинной частоты расстройства в общей популяции. Поэтому настоятельно рекомендуется, чтобы родители ребенка и, возможно, братья и сестры страдающего от СТК в связи с мутацией в генах TCOF1, POLR1B, POLR1C или POLR1D, были проверенны, даже если кажутся здоровыми. Это важно для будущего планирования семьи. Не следует предполагать, что мутация у больного ребенка произошла самопроизвольно, просто потому, что у родителей нет лицевых различий. Тем не менее, следует отметить, что у некоторых людей (приблизительно 10-15%) с особенностями и физическими признаками СТК нет мутаций ни в одном из четырех вышеупомянутых генов, что позволяет предположить, что дополнительные, еще не идентифицированные гены также могут вызывать СТК.
Диагностика
Диагноз СТК ставится на основании тщательной клинической оценки, подробного анамнеза пациента и определения характерных физических признаков. При рождении присутствуют многие сопутствующие аномалии, такие как пороки развития или отсутствие наружного уха.
— Клиническое тестирование и обследование.
Специализированные рентгенологические исследования подтвердят наличие и/или степень определенных наблюдаемых черепно-лицевых аномалий. Например, такие визуальные исследования показывают аномальную малость челюсти (микрогнатия) из-за недоразвития кости нижней челюсти (гипоплазия нижней челюсти), наличие и/или степень гипоплазии, затрагивающей определенные части черепа, и/или наличие дополнительных пороков развития уха, которые нельзя увидеть во время клинической оценки.
Кроме того, у тех больных, у которых наблюдается мало признаков, тщательное клиническое обследование и рентгенография черепно-лицевой области могут продемонстрировать едва различимое присутствие определенных характерных признаков (например, гипоплазию скуловых дуг), связанных с синдромом. Поскольку синдром Тричера-Коллинза обладает несколькими физическими особенностями, которые могут встречаться при других черепно-лицевых синдромах, многие исследователи рекомендуют делать диагностическое подтверждение посредством молекулярно-генетического тестирования и/или, в некоторых случаях, тщательной, детальной семейной истории.
Молекулярно-генетическое тестирование для подтверждения диагноза доступно в коммерческих и академических исследовательских лабораториях для выявления мутаций в генах TCOF1, POLR1B, POLR1C и POLR1D. Приблизительно 80% людей имеют идентифицируемую мутацию гена TCOF1. Кроме того, генетическое подтверждение TCOF1, POLR1B, POLR1C или POLR1D мутаций может быть обнаружена до рождения (пренатально) путем амниоцентеза и взятия проб ворсинчатого хориона, если мутация была выявлена у больного члена семьи. В некоторых случаях ультразвуковое исследование плода, в котором используются отраженные звуковые волны для создания изображения развивающегося плода, может выявить характерные признаки, указывающие на наличие СТК у ребенка. Родственники, особенно родители и братья или сестры, лиц с диагнозом СТК должны быть тщательно обследованы, поскольку легкие случаи часто остаются нераспознанными и не диагностируемыми.
Стандартные методы лечения
От синдрома Тричера-Коллинза нет никакого лекарства. Лечение направлено на конкретные симптомы, которые проявляются у каждого человека. Лечение может потребовать скоординированных усилий группы специалистов. Педиатры, специалисты по лечению уха, носа и горла (детские отоларингологи), детские стоматологи, медсестры, пластические хирурги, логопеды, аудиологи, офтальмологи, психологи, генетики и другие медицинские работники может потребоваться систематическое и всестороннее планирование лечения ребенка, затронутого болезнью.
Врачи регулярно проводят мониторинг лиц с СТК для выявления определенных отклонений, которые могут быть связаны с расстройством. Например, следует внимательно следить за слухом больного, страдающего от СТК, чтобы выявить любые признаки потери слуха. Оценка слуха младенца имеет решающее значение, и полную оценку следует проводить в раннем возрасте, даже до одного года, а затем ежегодно, чтобы обеспечить правильное развитие речи.
Для визуализации внутренней части глаза используется инструмент (офтальмоскоп), позволяющий обнаружить любое возможное ухудшение зрения. Этот осмотр важен для обеспечения соответствующих профилактических мер и/или оперативного лечения больных, у которых наблюдаются отклонения в глазах в связи с синдромом Тричера-Коллинза (например, колобомы, косоглазия, микрофтальмии). Пострадавшие люди также должны находиться под наблюдением на предмет патологий челюсти и зубов.
Раннее вмешательство важно для обеспечения того, чтобы затронутые дети раскрыли свой потенциал. Специальные услуги, которые могут быть полезными, включают логопедическую, специальную социальную поддержку и другие медицинские, социальные и/или профессиональные услуги.
Прогноз
Обычно люди с синдромом Тричера-Коллинза становятся полноценными взрослыми с нормальным интеллектом. При правильном обращении и лечении ребенка ожидаемая продолжительность жизни примерно такая же, как и у населения в целом. В некоторых случаях прогноз зависит от конкретных симптомов и тяжести больного. Например, очень тяжелые случаи СТК могут вызвать перинатальную смерть из-за нарушения дыхательных путей.
- 2.1 TCOF1
- 2.2 Другие мутации
- 3.1 Генетическое консультирование
- 3.2 Пренатальная диагностика
- 3.3 Клинические данные
- 3.4 Рентгенограммы
- 3.5 КТ
- 3.6 Дифференциальный диагноз
- 4.1 Потеря слуха
- 4.2 Психиатрия
Признаки и симптомы

Тот же ребенок, показанный спереди вверху в информационном окне, теперь виден сбоку, с маленькими ушами и подбородком, который находится далеко позади.
Симптомы у людей с синдромом Тричера Коллинза различаются. У некоторых людей поражение настолько легкое, что их диагноз остается невыявленным, в то время как у других наблюдается умеренное или тяжелое поражение лица и опасные для жизни нарушения дыхательных путей. Большинство признаков ТКС симметричны и распознаются уже при рождении.
Наиболее частым симптомом синдрома Тричера-Коллинза является недоразвитие нижней челюсти и скуловой кости.. Это может сопровождаться втягиванием язычка. Маленькая нижняя челюсть может привести к плохой окклюзии зубов или, в более тяжелых случаях, затрудненному дыханию или глотанию. Дыхательная система ребенка с синдромом Тричера Коллинза является основной проблемой, когда ребенок рождается, и другие проблемы решаются после того, как будут решены респираторные проблемы. Недоразвитие скуловой кости придает щекам запавший вид.
Наружное ухо иногда маленькое, повернуто, деформировано или полностью отсутствует у людей с ТКШ. Также описывается симметричное двустороннее сужение или отсутствие наружного слухового прохода. В большинстве случаев кости среднего уха и полость среднего уха деформированы. Пороки развития внутреннего уха описываются редко. В результате этих аномалий у большинства людей с TCS наблюдается кондуктивная потеря слуха.
У большинства людей также возникают проблемы со зрением, в том числе колобомы (выемки) в нижних веках, частичные или частичные. полное отсутствие ресниц на нижнем веке, веки, наклоненные вниз, опущение верхнего и нижнего века и сужение слезных протоков. Может возникнуть потеря зрения, связанная с косоглазием, аномалиями рефракции и анизометропией. Это также может быть вызвано сильной сухостью глаз, следствием аномалий нижних век и частых глазных инфекций.
Хотя череп неправильной формы не характерен для синдрома Тричера Коллинза, брахицефалия с битемпоральным сужением иногда наблюдается. Расщелина неба также является обычным явлением.
Стоматологические аномалии наблюдаются у 60% пострадавших людей, включая агенез зубов (33%), изменение цвета (помутнение эмали) (20%), неправильное смещение зуба. первые моляры верхней челюсти (13%) и большое расстояние между зубами. В некоторых случаях аномалии зубов в сочетании с гипоплазией нижней челюсти приводят к неправильному прикусу. Это может привести к проблемам с приемом пищи и невозможностью закрыть рот.
Менее распространенные признаки TCS могут усугубить проблемы с дыханием у пострадавшего человека, включая апноэ во сне. Атрезия хоан или стеноз — это сужение или отсутствие хоан, внутреннего отверстия носовых ходов, которое также может наблюдаться. Недоразвитие глотки также может сужать дыхательные пути.
Реже встречаются признаки, связанные с TCS, включая деформации носа, высокое арочное небо, макростомию, преаурикулярные волосы смещение, волчья пасть, гипертелоризм, выемка на верхнем веке и врожденные пороки сердца.
Хотя деформация лица часто связана с задержкой развития и умственной отсталостью, более 95% людей, страдающих TCS, имеют нормальный интеллект. Психологические и социальные проблемы, связанные с лицевой деформацией, могут повлиять на качество жизни людей с СКС.
Генетика
Синдром Тричера Коллинза наследуется по аутосомно-доминантной модели.
Мутации в генах TCOF1, POLR1C или POLR1D могут вызывать синдром Тричера Коллинза. Мутации гена TCOF1 являются наиболее частой причиной заболевания, а мутации генов POLR1C и POLR1D вызывают еще 2% случаев. У людей без идентифицированной мутации в одном из этих генов генетическая причина состояния неизвестна. Гены TCOF1, POLR1C и POLR1D кодируют белки, которые играют важную роль в раннем развитии костей и других тканей лица. Мутации в этих генах снижают производство рРНК, что может вызвать самоуничтожение (апоптоз) определенных клеток, участвующих в развитии костей и тканей лица. Непонятно, почему эффекты снижения рРНК ограничиваются развитием лица. Мутации в TCOF1 и POLR1D вызывают аутосомно-доминантную форму Treacher Collins, а мутации в POLR1C вызывают аутосомно-рецессивную форму.
TCOF1
TCOF1 является первичным геном, связанным с TCS; мутация в этом гене обнаруживается у 90-95% людей с TCS. Однако у некоторых людей с типичными симптомами TCS мутации в TCOF1 не обнаружены. Исследование ДНК привело к идентификации мутаций, обнаруженных в TCOF1. Большинство мутаций представляют собой небольшие делеции или вставки, хотя также были идентифицированы сайт сплайсинга и миссенс-мутации.
Анализ мутаций выявил более 100 болезнетворных мутаций в TCOF1, которые в основном являются семейно-специфическими мутациями. На единственную рецидивирующую мутацию приходится около 17% случаев.
TCOF1 обнаруживается на 5-й хромосоме в области 5q32. Он кодирует относительно простой ядрышковый белок, называемый патока, который, как считается, участвует в сборке рибосом. Мутации в TCOF1 приводят к гаплонедостаточности белка патоки. Гаплонедостаточность возникает, когда диплоидный организм имеет только одну функциональную копию гена, потому что другая копия инактивирована мутацией. Одна нормальная копия гена не производит достаточного количества белка, что вызывает болезнь. Гаплонедостаточность белка патоки приводит к истощению предшественника клеток нервного гребня, что приводит к уменьшению количества клеток гребня, мигрирующих в первую и вторую глоточные дуги. Эти клетки играют важную роль в развитии черепно-лицевого внешнего вида, и потеря одной копии патоки влияет на способность клеток формировать кости и ткани лица.
Другие мутации
Мутации POLR1C и POLR1D ответственны за меньшую часть случаев Treacher Collins. POLR1C находится на хромосоме 6 в положении 6q21.2, а POLR1D находится на хромосоме 13 в положении 13q12.2. Эти гены кодируют белковые субъединицы, общие для РНК-полимеразы I и III. Обе эти полимеразы важны для биогенеза рибосом.
Диагноз
Генетическое консультирование
TCS наследуется аутосомно-доминантным способом и пенетрантностью пораженный ген почти завершен. Однако некоторые недавние исследования описали некоторые редкие случаи, когда пенетрантность в TCS не была полной. Причинами могут быть переменная экспрессия, неполная пенетрантность или мозаицизм зародышевой линии. Только 40% мутаций передаются по наследству. Остальные 60% являются результатом мутации de novo, когда ребенок имеет новую мутацию в ответственном гене и не унаследовал ее ни от одного из родителей. В исходе болезни возникает межсемейная и внутрисемейная изменчивость. Это говорит о том, что при рождении пораженного ребенка важно обследовать родителей, чтобы определить, присутствует ли пораженный ген, поскольку у родителя может быть легкая форма заболевания, которая не была диагностирована. В этом случае риск рождения еще одного больного ребенка составляет 50%. Если у родителей нет пораженного гена, риск рецидива низок. В следующих поколениях тяжесть клинических симптомов увеличивается.
Пренатальная диагностика
Мутации в основных генах, ответственных за TCS, могут быть обнаружены с помощью взятия проб ворсинок хориона или амниоцентеза. Эти методы не позволяют обнаружить редкие мутации. Ультрасонография может использоваться для выявления черепно-лицевых аномалий на более поздних сроках беременности, но может не выявить более легкие случаи.
Клинические данные
TCS часто сначала подозревают при наличии характерных симптомов, наблюдаемых во время физического осмотра. Однако клинические проявления СТК могут напоминать другие заболевания, что затрудняет диагностику. Классификация OMENS была разработана как комплексный и поэтапный подход к дифференциации заболеваний. Этот акроним описывает пять различных проявлений дисморфологии, а именно орбитальную асимметрию, гипоплазию нижней челюсти, деформацию ушной раковины, развитие нервов и заболевание мягких тканей.
- O0: нормальный размер орбиты, положение
- O1: аномальный размер орбиты
- O2: аномальное положение орбиты
- O3: аномальный размер и положение орбиты
- M0: нормальная нижняя челюсть
- M1: маленькая нижняя челюсть и гленоид ямка с короткой ветвью
- M2: ветвь короткая и неправильной формы
- 2A: гленоидная ямка в анатомически приемлемом положении
- 2B: височно-нижнечелюстной сустав снизу (ВНЧС), медиально, смещенный кпереди, с сильно гипоплазированным мыщелком
- M3: полное отсутствие ветви, суставной ямки и височно-нижнечелюстного сустава
- E0: нормальное ухо
- E1: незначительная гипоплазия и купирование всех структур
- E2: Отсутствие наружного слухового прохода с различной гипоплазией ушной раковины
- E3: Неправильное положение дольки с отсутствием ушной раковины, дольчатый остаток обычно нижний смещенный кпереди
- N0: нет лицевой нерв вовлечение
- N1: поражение верхнего лицевого нерва (височные или скуловые ветви)
- N2: Поражение нижнего лицевого нерва (буккального, нижнечелюстного или шейного)
- N3: поражены все ветви
- S0: нет дефицита мягких тканей или мышц
- S1: минимальное количество тканей или мышц дефицит
- S2: умеренный тканевый или мышечный дефицит
- S3: тяжелый тканевый или мышечный дефицит
Рентгенограммы
Для подтверждения диагноза в TCS используется несколько методов. 99>
ортопантомограмма (OPG) — это панорамный рентгеновский снимок верхней и нижней челюсти. Он показывает двухмерное изображение от уха до уха. В частности, OPG способствует точному послеоперационному наблюдению и мониторингу роста костей при лечении с использованием моно- или двойного дистрактора. Таким образом, некоторые особенности TCS можно увидеть на OPG, но используются более совершенные методы, позволяющие охватить весь спектр аномалий TCS вместо того, чтобы показывать только аномалии челюсти.
Другой метод радиографической оценки — получение рентгеновского изображения всей головы. Боковая цефалометрическая рентгенограмма в TCS показывает гипоплазию лицевых костей, таких как скуловая кость, нижняя челюсть и сосцевидный отросток.
Наконец, затылочные рентгенограммы используются для обнаружения гипоплазии или нарушения целостности скуловой дуги.
КТ височной кости с использованием тонких срезов позволяет диагностировать степень стеноза и атрезии наружного слухового прохода, состояние полости среднего уха, отсутствие или диспластические и рудиментарные косточки, или аномалии внутреннего уха, такие как недостаточная улитка. Двух- и трехмерные КТ-реконструкции с VRT и покрытием костей и кожи полезны для более точной постановки и трехмерного планирования реконструктивной хирургии нижней челюсти и наружного уха.
Дифференциальный диагноз
Другие болезни имеют сходные характеристики с синдромом Тричера Коллинза. В дифференциальной диагностике следует учитывать акрофациальные дизостозы. Внешний вид лица напоминает синдром Тричера Коллинза, но у этих людей возникают дополнительные аномалии конечностей. Примерами этих заболеваний являются синдром Нагера и синдром Миллера.
При дифференциальной диагностике также следует учитывать окулоаурикуловертебральный спектр. Примером является гемифациальная микросомия, которая в первую очередь влияет на развитие уха, рта и нижней челюсти. Эта аномалия может возникать двусторонне. Еще одно заболевание, относящееся к этому спектру, — синдром Гольденхара, который включает в себя позвоночные аномалии, эпибульбарные дермоиды и деформации лица.
Лечение
Лечение людей с TCS может включать вмешательство профессионалов из разных дисциплин. В первую очередь заботятся о дыхании и кормлении из-за гипоплазии нижней челюсти и обструкции гортани языком. Иногда им может потребоваться трахеостомия для поддержания проходимости дыхательных путей и гастростомия для обеспечения адекватного потребления калорий и защиты дыхательных путей. Корректирующая хирургия лица проводится в определенный возраст, в зависимости от состояния развития.
Обзор настоящих рекомендаций:
- Если присутствует волчья пасть, восстановление обычно требует место в возрасте 9–12 месяцев. Перед операцией необходима полисомнография с установленной небной пластиной. Это может предсказать послеоперационную ситуацию и дает представление о вероятности наличия апноэ во сне (СОАС) после операции.
- Для лечения потери слуха используется усиление костной проводимости, логопедия и образовательное вмешательство во избежание языковых / речевых проблем. слуховой аппарат с костной фиксацией — альтернатива для людей с аномалиями уха.
- Скуловая и орбитальная реконструкция выполняется, когда краниоорбитозигоматическая кость полностью сформирована, обычно в возрасте от 5 до 5 лет. 7 лет. У детей чаще всего используют аутотрансплантат. В сочетании с этой трансплантацией можно использовать липофилинг в периорбитальной области для получения оптимального результата реконструкции. Реконструкция нижнего века колобома включает использование кожно-мышечного лоскута, который приподнимается и таким образом закрывает дефект века.
- Реконструкция наружного уха обычно выполняется, когда человеку не менее восемь лет. Иногда можно лечить наружный слуховой проход или среднее ухо.
- Оптимальный возраст для проведения реконструкции челюстно-нижнечелюстной кости является спорным; по состоянию на 2004 год использовалась эта классификация:
- Тип I (легкая форма) и Тип IIa (умеренная) 13–16 лет
- Тип IIb (порок развития средней и тяжелой степени) при зрелости скелета
- Тип III (тяжелый) 6–10 лет
- Когда зубы режутся, зубы должны находиться под наблюдением ортодонта, чтобы убедиться в отсутствии аномалий. При обнаружении таких аномалий, как вывих или чрезмерное разрастание зубов, соответствующие меры могут быть предприняты как можно скорее.
- Ортогнатическое лечение обычно проводится после 16 лет; к этому моменту все зубы встали на свои места, а челюсти и протезы стали зрелыми. При обнаружении СОАС уровень обструкции определяется с помощью эндоскопии верхних дыхательных путей. Выдвижение нижней челюсти может быть эффективным способом улучшить как дыхание, так и эстетику, в то время как пластика подбородка только восстанавливает его профиль.
- Если реконструкция носа необходима, она обычно выполняется после ортогнатической операции и в возрасте старше 18 лет..
- Контур мягких тканей лица обычно требует коррекции в более позднем возрасте из-за зрелости лицевого скелета. Использование микрохирургических методов, таких как перенос свободного лоскута, улучшило коррекцию контуров мягких тканей лица. Еще один метод улучшения контуров мягких тканей лица — липофилинг. Например, липофилинг используется для реконструкции век.
Потеря слуха
Потеря слуха при синдроме Тричера Коллинза вызвана деформацией структур в наружном и среднем ухе. Потеря слуха обычно двусторонняя с кондуктивной потерей около 50-70 дБ. Даже в случаях с нормальными ушными раковинами и открытыми наружными слуховыми проходами цепочка слуховых косточек часто деформируется.
Попытки хирургической реконструкции наружного слухового прохода и улучшения слуха у детей с СКС не дали положительных результатов.
Слуховая реабилитация с помощью слуховых аппаратов с костной фиксацией (BAHA) или обычных средств костной проводимости оказалась предпочтительнее хирургической реконструкции.
Психиатрические
Заболевание может быть ассоциировано с рядом психологических симптомов, включая тревогу, депрессию, социальную фобию и беспокойство по поводу образа тела; люди также могут подвергаться дискриминации, издевательствам и обзываниям, особенно в молодом возрасте. Эти вопросы должны учитывать многопрофильная команда и родительская поддержка.
Эпидемиология
TCS встречается примерно у одного из 50 000 новорожденных в Европе. По оценкам, во всем мире он встречается от одного из 10 000 до 1 из 50 000 рождений.
История
Синдром назван в честь Эдварда Тричера Коллинза (1862–1932), англичанин хирург и офтальмолог, которые описали его основные черты в 1900 году. В 1949 году Адольф Франческетти и Дэвид Кляйн описали то же заболевание на основании своих собственных наблюдений как нижнечелюстно-лицевой дизостоз.. Термин нижнечелюстно-лицевой дизостоз используется для описания клинических проявлений.
Культура
Статья в New York Times, июль 1977 г. , перепечатанная в многочисленных газетах по всей стране в течение следующих недель. эта болезнь впервые привлекла внимание многих людей.
Беспорядок был показан в шоу Nip / Tuck, в эпизоде «Blu Mondae ». TLC «Рожденные без лица» с Джулианой Ветмор, которая родилась с самым тяжелым случаем в истории болезни этого синдрома, у которой отсутствуют 30-40% костей лица.
В 2010 году BBC Three документальный фильм Люби меня, люби мое лицо, освещал дело человека, Джоно Ланкастера, с этим состоянием. В 2011 году BBC Three вернулся к Джоно, чтобы осветить его и его партнершу Лору стремление создать семью в фильме «Так что, если мой ребенок родился как я?», Который впервые вышел в эфир в рамках сезона программ BBC Three о воспитании детей. Первый фильм был показан на BBC One незадолго до первой трансляции второго фильма на BBC Three. Третий фильм Ланкастера BBC Three, «В поисках семьи» на Facebook, в котором рассматривается усыновление, вышел в эфир в 2011 году.
В Wonder, детский роман, главный герой — ребенок с синдромом Тричера Коллинза. В ноябре 2017 года вышла экранизация 2017 года с Джулией Робертс, Оуэном Уилсоном и Джейкобом Тремблеем. Элисон Мидстокк, актриса и модель с этим заболеванием.
Синдром Тричера-Коллинза-Франческетти (мандибуло-фациальный дизостоз). Поиск мутаций в гене TCOF1, м. (Treacher-Collins Syndrome, Franceschetti-Klein Syndrome, Mandibulofacial Dysostosis without Limb Anomalies, Gene TCOF1, Mut.)
Синдром Тричер – Коллинза — Франческетти (мандибулофациальный дизостоз, TCOF, OMIM154500) — аутосомно-доминантное заболевание, характеризующееся нарушением черепно-лицевого развития. Клинические признаки: антимонголоидный разрез глаз, колобома (дефект) нижних век, микрогнатия (гипоплазия нижней челюсти), двусторонняя гипоплазия скуловых костей и орбит, аномалии ушных раковин, дефект слухового прохода, приводящий к кондуктивной тугоухости. Часто встречается высокое арковидное нёбо или расщелина нёба, макростомия (большая ротовая щель), отсутствие ресниц на нижнем веке. В некоторых случаях отмечаются рост волос на щеках, колобомы верхнего века и радужки. У большинства пациентов слаборазвитые лицевые кости, что приводит к «затонувшему» лицу: крупный нос и очень маленькие челюсти и подбородок (микрогнатия). У некоторых больных присутствует волчья пасть. При тяжёлой патологии микрогнатия может вытеснять язык у новорождённых, создавая опасную преграду в ротоглотке для дыхания.
Для заболевания характерна высокая пенетрантность (т.е. высокая вероятность проявления признаков болезни у людей с мутацией) и различная экспрессивность (т.е. различный характер и тяжесть проявления болезни). Молекулярно-генетической причиной заболевания являются, как правило, нонсенс мутации в генеTCOF1, приводящие к возникновению преждевременного стоп кодона и, как следствие, к гаплонедостаточности (состояние, при котором половинного количества генного продукта недостаточно для нормального функционирования организма). Продукт гена — ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. Возможна прямая молекулярно-генетическая диагностика этого синдрома, которая заключается в выявлении изменений нуклеотидной последовательности в гене TCOF1 методом прямого автоматического секвенирования.
Аутосомно-доминантный. Для заболевания характерна высокая пенетрантность (т.е. высокая вероятность проявления признаков болезни у людей с мутацией) и различная экспрессивность (т.е. различный характер и тяжесть проявления болезни).
Мутацию в гене TCOF1 можно выявить в 78%-93% случаев, в 8% выявляют мутации в генах POLR1C или POLR1D.
Продукт гена — ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. У больных весьма характерное лицо. Клинические признаки: антимонголоидный разрез глаз, колобома (дефект) нижних век, микрогнатия (гипоплазия нижней челюсти), двусторонняя гипоплазия скуловых костей и орбит, аномалии ушных раковин, дефект слухового прохода, приводящий к кондуктивной тугоухости. Часто встречается высокое арковидное небо или расщелина неба, макростомия (большая ротовая щель), отсутствие ресниц на нижнем веке. В некоторых случаях отмечаются рост волос на щеках, колобомы верхнего века и радужки. Порок развития уха заключается в деформации ушной раковины, отсутствии костного отдела наружного слухового прохода, недоразвитии барабанной полости и слуховых косточек. Отмечаются также гипоплазия больших пальцев лучевой и локтевой костей, расщелины неба. Тугоухость смешанного характера с одновременным поражением звукопроведения и звуковосприятия.
Интеллект, как правило, не страдает. Трудности с дыханием и питанием могут возникнуть в первые годы из-за стеноза верхних дыхательных путей и ограниченного открытия рта.
Литература
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Кадышев В.В., Бессонова Л.А., Зинченко Р.А. Распространенность синдрома Тричер Коллинз-Франческетти в Кировской области . Медицинская генетика, 2009 г. Том 8. №12 (90) , 2009.
- OMIM.
Специальной подготовки к исследованию не требуется.
- анкета генетического исследования *;
- направительный бланк;
- информированное согласие.
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Республики Беларусь.
Литература
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Кадышев В.В., Бессонова Л.А., Зинченко Р.А. Распространенность синдрома Тричер Коллинз-Франческетти в Кировской области . Медицинская генетика, 2009 г. Том 8. №12 (90) , 2009.
- OMIM.
Типичная клиническая картина.
Литература
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Кадышев В.В., Бессонова Л.А., Зинченко Р.А. Распространенность синдрома Тричер Коллинз-Франческетти в Кировской области . Медицинская генетика, 2009 г. Том 8. №12 (90) , 2009.
- OMIM.
Интерпретация результатов исследования содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
синдром I-II жаберных дуг, гемифациальная микросомия, синдром Гольденхара, синдром Пьера Робена, синдромы Нагера.
- Мутация не выявлена.
- Мутация выявлена в гетерозиготном состоянии.
- Мутация выявлена в гомозиготном состоянии.
- Мутация выявлена в компаунд –гетерозиготном состоянии.
Литература
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Кадышев В.В., Бессонова Л.А., Зинченко Р.А. Распространенность синдрома Тричер Коллинз-Франческетти в Кировской области . Медицинская генетика, 2009 г. Том 8. №12 (90) , 2009.
- OMIM.
Сдать анализ «Синдром Тричера-Коллинза-Франческетти (мандибуло-фациальный дизостоз). Поиск мутаций в гене TCOF1, м. (Treacher-Collins Syndrome, Franceschetti-Klein Syndrome, Mandibulofacial Dysostosis without Limb Anomalies, Gene TCOF1, Mut.)» вы можете в Минске и других городах Республики Беларусь. Обратите внимание, что цена анализа, стоимость процедуры взятия биоматериала, методы и сроки выполнения исследований в региональных медицинских офисах могут отличаться
Синдром Тречер Коллинза или Франческетти — Клейна. Челюстно-лицевой дизостоз
Синдром Тречер Коллинза или Франческетти — Клейна. Челюстно-лицевой дизостоз
Хотя синдром был, возможно, впервые описан Thomson (1846— 1947), заявка на это открытие обычно отдается Berry или особенно Treacher Collins (имя обычно часто ошибочно связывают дефисом), который описал существенные компоненты синдрома. Franceschelti и Klein опубликовали обширный обзор, посвященный этому синдрому, и дали ему название челюстно-лнцевого дизостоза. Rogers произвел разносторонний анализ случаев. Всего опубликовано более чем 250 случаев.
Клинические данные. Данные осмотра. Внешний вид лица характерен. Косой опущенный вниз разрез глаз, запавшие кости щек, деформированная ушная раковина, отступающий назад подбородок (ретрогнатия) и большой, похожий на рыбий, рот так хорошо представляют клиническую картину болезни, что делают ее незабываемой. Другой необычной чертой является своеобразный рост волос в виде языка, распространяющегося в направлении щек (Gorlin et al.).
Несмотря на то что разрез глаз имеет косую, антимонголоидную форму, зрение обычно нормальное. Часто (примерно у 75% больных) имеется колобома в наружной трети нижнего века. Почти у 50% больных отмечается недостаток ресниц медиальнее колобомы.
Носолобный угол обычно закрыт, а переносье приподнято. Из-за недостаточного развития скуловых костей нос выглядит крупным. Ноздри нередко узки, а хрящ носа гипопластичен.
Нижняя челюсть почти всегда гипопластична, угол ее более тупой, чем в норме, а рама недоразвита. Под поверхностью тела нижней челюсти часто имеется резко выраженная впадина. Более чем у 40% больных отмечается высокое или расщепленное небо, а также недостаточное смыкание зубов.
Орган слуха. Ушная раковина часто деформирована, сморщена спереди или смещена. Наружный слуховой проход, когда он имеется, узкий и косой (Harrison). Около 85% больных имеют деформированную ушную раковину и более чем у 1/3 больных отсутствует наружный слуховой проход или отмечается дефект слуховых косточек с проводящей глухотой (Stovin et al.). Отмечен также нейросенсорный компонент глухоты (Kittel, Fleischer-Peters, Partsch, Hulse).
Рентгенологические исследования показали наличие склероза среднего и изредка внутреннего уха с плохо очерченными структурами. Слуховые косточки, а также улитка и вестибулярный аппарат могут отсутствовать или быть грубо деформированными (McKenzie, Craig, Pavsek, Herberts).
Хирургическое исследование выявило такие аномалии, как фиксацию молоточка, синостоз и уродство молоточка и наковальни, отсутствие стремени и овального окна, отсутствие сухожилия m. stapedius, деформацию наковальни и стремени, отсутствие наковальни, анкилоз основания стремени, костный мостик в канале лицевого нерва, а также полное отсутствие среднего уха и надбарабанного пространства. Полость среднего уха может быть заполнена соединительной тканью (Altmann, Plester, Holborow, Keerl). Plester, Keerl и Edwards обнаружили монолитное стремя и истончение длинного отростка наковальни.

Почти у 25% больных была обнаружена патология лабиринта (Stovin, Herberts). Между трагусом и углом рта могут встречаться внеушные бугорки или слепые свищи.
Вестибулярная система. Результаты исследований вестибулярной системы не опубликованы.
Лабораторные данные. Рентгенологически нижний край глазницы дефектен, а полость глазницы овальной формы с наклоненной вниз и наружу крышей. Тело скуловой кости может вовсе отсутствовать, по чаще, однако, оно значительно и симметрично недоразвито и не соединено со скуловыми дугами (нет височной ветви и фактически отсутствует верхнечелюстная ветвь). Сосцевидный отросток безвоздушен и. часто склерозирован. Параназальные синусы очень малы и могут полностью отсутствовать.
Томография височных костей показала атрезию наружного слухового прохода, значительное уменьшение размера барабанной полости, частую недостаточность верхнего отдела среднего уха, утолщение ушной капсулы, расположение дна средней ямки ниже уровня наружного полукружного канала и неправильное направление лицевого нерва (Terrahe).
Наследственность. Синдром наследуется по аутосомно-доминантному типу с неполной пенетрантностью и варьирующей экспрессивностью. Создается впечатление, что ген обладает летальным эффектом, так как в семьях часто наблюдаются выкидыши или ранняя постнатальиая смерть детей.
Диагноз. От челюстно-лицевого дизостоза необходимо отличать глазо-ушно-позвоночный синдром (окуло-аурикуло-вертебральный синдром, синдром Гольденхара, гемифациальная микросомия). Характерным для синдрома Гольденхара является асимметрия лица, односторонняя гипоплазия рамы нижней челюсти, деформированная ушная раковина. Менее часто наблюдаются эпибульбарная дермоидная киста, колобома верхнего века, ушные бугорки, макростомия, односторонний парез лицевого нерва и аномалии позвонков (Gorlin et al.). Наследование, вероятно, мультифакторнальное.
Лечение. Совместные усилия должны быть направлены на коррекцию микротии, гипоплазии нижней челюсти, песмыкаиия зубов, гипоплазии скуловых костей. К улучшению слуха обычно ведет тимпанотомия с введением слухового протеза.
Прогноз. Глухота не прогрессирует.
Выводы. Характеристика этого синдрома включает: 1) аутосомно-доминантное наследование с варьирующей экспрессивностью; 2) гипоплазию скуловых костей, в результате чего возникает антимонголоидный разрез глаз; 3) колобому нижнего века п отсутствие ресниц медиальнее колобомы; 4) гипоплазию нижней челюсти; 5) деформацию ушной раковины, наружного слухового прохода и структур среднего уха; 6) проводящую глухоту.
Синдром Тричера-Коллинза ( Мандибулофациальный дизостоз , Синдром Тричера Коллинза-Франческетти , Челюстно-лицевой дизостоз )
Синдром Тричера Коллинза – это генетическая (иногда наследственная) болезнь, сопровождающаяся деформациями костей и мягких тканей лица. К симптомам относятся грубые дефекты строения лица: антимонголоидный разрез глаз, вырезки ткани век (колобомы), уменьшенные размеры челюсти и скул, гипоплазия и аномалии структур уха, расщелина или арковидная форма неба, увеличенные размеры ротовой щели и языка, слаборазвитые кости лица. Диагноз устанавливается по данным клинического осмотра, биогенетического теста и семейного анамнеза. Лечение симптоматическое, направлено на улучшение слуха, устранение жизнеугрожающих деформаций и косметических дефектов хирургическим способом.
МКБ-10
Общие сведения
У синдрома Тричера Коллинза есть несколько синонимов: челюстно-лицевой дизостоз, синдром Тричера Коллинза-Франческетти, мандибулофациальный дизостоз. Впервые патологию описал офтальмолог из Великобритании Эдвард Тричер Коллинз в 1900 году, поэтому наиболее распространено название, соответствующее его имени. Обширный обзор заболевания был сделан в 1949 году европейскими исследователями Э. Франческетти и Д. Клейном. В настоящее время понятие «синдром Тричера Коллинза» более распространено в Великобритании и США, а термин «синдром Франческетти-Клейна» чаще используется в странах Европы. Эпидемиология болезни составляет 1:50 000. Среди мальчиков и девочек заболеваемость одинакова.
Причины
При мутациях в гене TCOF1 тип наследования синдрома аутосомно-доминантный с показателем пенетрантности 90%. Это означает, что при мутации в одной хромосоме из пары вероятность проявления болезни очень высока. У больного родителя риск рождения ребенка с синдромом Тричера Коллинза составляет 50%. Возможна наследственная передача дефекта и спорадические генетические изменения (новые мутации). Экспрессивность мутации переменная – в пределах одной семьи вероятно как ослабление, так и усиление симптомов заболевания у последующих поколений. При дефектах генов POLR1C и POLR1D наследование происходит по аутосомно-рецессивному типу. В парах, где родитель имеет синдром, вероятность рождения больного малыша составляет 25%.
Патогенез
Пятая хромосома ответственна за правильное формирование скелета в период внутриутробного развития. Локализованный в ней ген TCOF1 кодирует структуру и синтез ядерного транспортного белка Treacle. Данный протеин экспрессируется в большинстве тканей организма в эмбриональном и постэмбриональном периоде, участвует в переносе генетической информации с ДНК на РНК.
В основе синдрома чаще всего лежит нонсенс-мутация, приводящая к образованию преждевременного кодона терминации и развитию гаплонедостаточности – дефицита белка, необходимого для нормального формирования лицевой части черепа. Здоровый ген обеспечивает организм белком Treacle наполовину, но такого количества недостаточно для правильного развития лицевых структур. При изменениях в генах POLR1D и POLR1C процесс транскрипции ДНК нарушается из-за недостаточности фермента-катализатора ДНК-зависимой РНК-полимеразы. Клинические проявления синдрома такие же, как и при первичной недостаточности Treacle-протеина.
Симптомы
У больных наблюдаются аномалии в строении лица. Распространенным признаком, встречающимся в 80% случаев, является двусторонняя симметричная гипоплазия скуловых костей, инфраорбитального края и нижней челюсти. Внешне это проявляется своеобразным уплощенным бесформенным лицом, на котором выделяется нос, а остальные части «утоплены» в мягких тканях. Деформация челюсти обуславливает нарушение прикуса, формирование ортогнатии (постоянно приоткрытого рта). 89% больных имеют ограниченную возможность открывания рта и антимонголоидный тип разреза глаз с заметным опущением внешнего уголка. Данные особенности частично обусловлены патологическим строением височно-нижнечелюстного сустава.
У 69% пациентов определяется колобома радужки и нижних век в промежутке между средней и внешней третью, чаще она имеет треугольную форму. Ресницы на внешнем крае нижнего века отсутствуют. Небо арковидной формы, иногда сформирована расщелина (у 28% больных). Аномалии наружного уха представлены недоразвитием или полным отсутствием ушной раковины (микротией, анотией), атрезией наружного слухового прохода и деформацией слуховых косточек. Зачастую пациенты имеют кондуктивную тугоухость. В редких случаях диагностируется энхондрома, предкозелковые фистулы, аномальное строение сердца и позвоночника.
Осложнения
Микрогнатия и стеноз верхних дыхательных путей уже в первые годы жизни могут спровоцировать проблемы при приеме пищи и трудности дыхания вплоть до удушья. Своевременная диагностика заболевания позволяет спрогнозировать эти осложнения и предпринять меры по их предупреждению. Как правило, пациенты не имеют врожденных интеллектуальных расстройств, но при отсутствии коррекции нарушений слуха становится невозможным правильное формирование речи и обучение в обычных условиях. Дети начинают отставать в умственном развитии от сверстников, имеют задержку психического развития различной степени выраженности. В связи с наличием дефектов внешности и негативным отношением окружающих больные всех возрастов относятся к группе риска по возникновению депрессии, ипохондрии, тревожности и иных невротических расстройств.
Диагностика
Диагноз может быть установлен во время беременности или сразу после рождения. Обследование показано женщинам из группы риска и детям с врожденными лицевыми деформациями. В процессе диагностики принимают участие врачи-генетики и педиатры. Синдром Тричера-Коллинза необходимо дифференцировать с другими генетическими заболеваниями, при которых существует деформация лицевой части черепа, например, с синдромом Нагера и синдромом Гольденхара. Используются следующие методы:
Дополнительно назначаются обследования, позволяющие своевременно обнаружить жизнеугрожающие состояния, оценить степень деформации костей черепа. Определяется эффективность кормления ребенка, уровень насыщения гемоглобина кислородом, ритмичность и глубина дыхания. Для диагностики сохранности слуха на 5-6 день жизни проводится аудиологическое тестирование. Инструментальная диагностика включает рентгенографию черепа, КТ и МРТ головного мозга.
Лечение синдрома
Специфической терапии не существует. Лечение нацелено на устранение симптомов и последствий заболевания, предполагает проведение хирургических операций и реабилитационных мероприятий. Объем процедур и сроки их выполнения устанавливаются индивидуально с учетом наличия угрозы для жизни больного, противопоказаний и рисков, связанных с оперативным вмешательством. Общая схема лечения включает:
- Восстановление глотания и дыхания. При развитии респираторного дистресс-синдрома осуществляется трахеостомия, дистракция подвижной челюсти, неинвазивная вентиляция легких. При невозможности потребления пищи устанавливается гастростома.
- Восстановление слуха. Деформация наружного и среднего уха устраняется хирургическим путем, но потеря слуха чаще обусловлена повреждением слуховых мелких косточек, поэтому оперативные вмешательства с целью устранения тугоухости неэффективны. Предпочтительна реабилитация слуховыми аппаратами.
- Устранение внешних дефектов. Деформации корректируются методами пластической и нижнечелюстно-лицевой хирургии. Применяется липоскульптурирование, хирургическая дистракция костей, установка трансплантатов и хирургическое восстановление неба.
Прогноз и профилактика
Комплексное лечение и реабилитация значительно улучшают качество жизни больных. При легкой и умеренной выраженности синдрома прогноз благоприятный. Профилактика затруднена, поскольку заболевание является генетическим, а мутации способны возникать спонтанно. Супружеским парам, в которых один родитель болен, необходимо медико-генетическое консультирование и перинатальная диагностика синдрома на ранних сроках беременности. Для снижения риска вынашивания больного ребенка рекомендуется процедура экстракорпорального оплодотворения с предварительным отбором генетически здоровых эмбрионов.
1. Медицинская и клиническая генетика для стоматологов: учебник для вузов. Под ред. О. О. Янушевича – 2009.
2. Особенности стоматологической патологии при некоторых наследственных заболеваниях / Шишкова О.В., Максимова Ю.В. // Медицина и образование в Сибири – 2007 — №3.
Случай ранней пренатальной диагностики синдрома Тричера Коллинза (Treacher Collins syndrome, OMIM: 154500) 1-й тип, семейная форма

Доступная эффективность. Универсальный ультразвуковой сканер, компактный дизайн и инновационные возможности.
Синдром Тричера Коллинза (СТК, Treacher Collins syndrome) – это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза–Франческетти, синдром Франческетти–Цвалена–Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер – имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза–Франческетти [4, 5].
Популяционная частота СТК оценивается как 1:50 000 живорожденных [1, 2], однако некоторые авторы называют более частую встречаемость этого синдрома: 1:10 000 [6]. Больные легко узнаваемы, их можно нередко встретить на улицах, увидеть в социальных сетях и, иногда, на телеэкранах. В 2017 г. вышла кинокартина режиссера Стивена Чбоски с Джулией Робертс в главной роли, которая называется «Чудо», где рассказана история мальчика Огги Пулмана с синдромом Тричера Коллинза и прекрасно продемонстрирована вся сложность социальной адаптации таких детей.

Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса [1–6], также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости [1–8]. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев – это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 – q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4–6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип – мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Несмотря на давно описанный в литературе и хорошо известный врачам-генетикам диагноз, количество статей, посвященных случаям дородовой диагностики СТК, весьма ограничено. Это связано с трудностью визуализации и объективизации некоторых классических фенотипических признаков синдрома при проведении пренатальной эхографии [12]. Ультразвуковые проявления изменений лицевого фенотипа у плодов бывают не очевидны, и часто рождение таких детей является полной неожиданностью не только для их родителей, но и для врачей пренатальной диагностики. Явные после рождения «ядерные» признаки СТК, такие как гипоплазия скуловых костей, микрогнатия, расщелина нёба, колобома нижнего века, антимонголоидный разрез глаз, отсутствие ресниц, чаще всего остаются незамеченными, даже при современных возможностях ультразвуковых приборов, особенно когда нет генетической настороженности при осмотре, что бывает при возникновении мутации de novo у фенотипически здоровых родителей. Часто в пренатальном периоде могут наблюдаться многоводие и задержка роста плода [14, 15]. Внедрение в клиническую практику современных режимов сканирования при помощи объемной визуализации лицевого фенотипа значимо облегчает диагностику [16]. Положение глазных щелей, аномальная форма носа, низко расположенные уши – все эти хорошо известные основные признаки СТК очень сложно уверенно визуализировать в обычном рутинном 2D-режиме, но при применении 3D-технологий их дефиниция становится более очевидной [16, 17].
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
- Синдром Гольденхара. Изменения лица при синдроме Гольденхара почти всегда односторонние, асимметричные, включают в себя колобому верхнего, а не нижнего века, а также эпибульбарные дермоиды, преаурикулярные привески. При синдроме Гольденхара могут встречаться аномалии позвоночника и пороки сердца.
- Синдром Нагера. Фенотипически похож на СТК, однако для него характерны преаксиальные (со стороны большого пальца кисти) дефекты верхней конечности – редукционные пороки верхних конечностей (в диапазоне от гипоплазии до аплазии большого пальца с или без вовлечения лучевой кости).
- Синдром Миллера, известный как постаксиальный акрофациальный дизостоз. Характеризуется микрогнатией, расщелиной губы, различными аномалиями позвонков и сколиозом. Типичными признаками являются постаксиальные (со стороны мизинца кисти) пороки верхней конечности либо только мизинца.
- Синдром Пьера Робена характеризуется изолированной гипоплазией нижней челюсти, глоссоптозом, расщелиной нёба.
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким – 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12–13 нед беременности.
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2–4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).

Рис. 2. Микрогнатия — сагиттальный скан в 2D, беременность 13 нед.
Читайте также:
- Установка имплантируемого кардиовертера-дефибриллятора (ИКД)
- Гипертония у детей
- Средний отит
- Лучевые признаки меланомы вульвы и влагалища
- Метаболический алкалоз

УЗИ аппарат HM70A
Экспертный класс по доступной цене. Монокристальные датчики, полноэкранный режим отображения, эластография, 3D/4D в корпусе ноутбука. Гибкая трансформация в стационарный сканер при наличии тележки.
Синдром Тричера Коллинза (СТК, Treacher Collins syndrome) – это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза–Франческетти, синдром Франческетти–Цвалена–Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер – имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза–Франческетти [4, 5].
Популяционная частота СТК оценивается как 1:50 000 живорожденных [1, 2], однако некоторые авторы называют более частую встречаемость этого синдрома: 1:10 000 [6]. Больные легко узнаваемы, их можно нередко встретить на улицах, увидеть в социальных сетях и, иногда, на телеэкранах. В 2017 г. вышла кинокартина режиссера Стивена Чбоски с Джулией Робертс в главной роли, которая называется «Чудо», где рассказана история мальчика Огги Пулмана с синдромом Тричера Коллинза и прекрасно продемонстрирована вся сложность социальной адаптации таких детей.
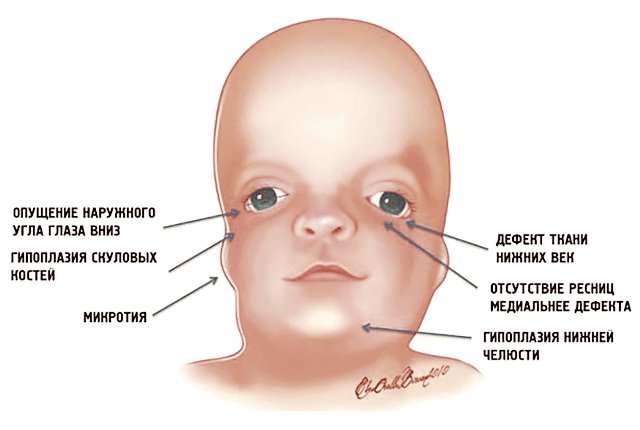
Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса [1–6], также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости [1–8]. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев – это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 – q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4–6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип – мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Несмотря на давно описанный в литературе и хорошо известный врачам-генетикам диагноз, количество статей, посвященных случаям дородовой диагностики СТК, весьма ограничено. Это связано с трудностью визуализации и объективизации некоторых классических фенотипических признаков синдрома при проведении пренатальной эхографии [12]. Ультразвуковые проявления изменений лицевого фенотипа у плодов бывают не очевидны, и часто рождение таких детей является полной неожиданностью не только для их родителей, но и для врачей пренатальной диагностики. Явные после рождения «ядерные» признаки СТК, такие как гипоплазия скуловых костей, микрогнатия, расщелина нёба, колобома нижнего века, антимонголоидный разрез глаз, отсутствие ресниц, чаще всего остаются незамеченными, даже при современных возможностях ультразвуковых приборов, особенно когда нет генетической настороженности при осмотре, что бывает при возникновении мутации de novo у фенотипически здоровых родителей. Часто в пренатальном периоде могут наблюдаться многоводие и задержка роста плода [14, 15]. Внедрение в клиническую практику современных режимов сканирования при помощи объемной визуализации лицевого фенотипа значимо облегчает диагностику [16]. Положение глазных щелей, аномальная форма носа, низко расположенные уши – все эти хорошо известные основные признаки СТК очень сложно уверенно визуализировать в обычном рутинном 2D-режиме, но при применении 3D-технологий их дефиниция становится более очевидной [16, 17].
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
- Синдром Гольденхара. Изменения лица при синдроме Гольденхара почти всегда односторонние, асимметричные, включают в себя колобому верхнего, а не нижнего века, а также эпибульбарные дермоиды, преаурикулярные привески. При синдроме Гольденхара могут встречаться аномалии позвоночника и пороки сердца.
- Синдром Нагера. Фенотипически похож на СТК, однако для него характерны преаксиальные (со стороны большого пальца кисти) дефекты верхней конечности – редукционные пороки верхних конечностей (в диапазоне от гипоплазии до аплазии большого пальца с или без вовлечения лучевой кости).
- Синдром Миллера, известный как постаксиальный акрофациальный дизостоз. Характеризуется микрогнатией, расщелиной губы, различными аномалиями позвонков и сколиозом. Типичными признаками являются постаксиальные (со стороны мизинца кисти) пороки верхней конечности либо только мизинца.
- Синдром Пьера Робена характеризуется изолированной гипоплазией нижней челюсти, глоссоптозом, расщелиной нёба.
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким – 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12–13 нед беременности.
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2–4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).
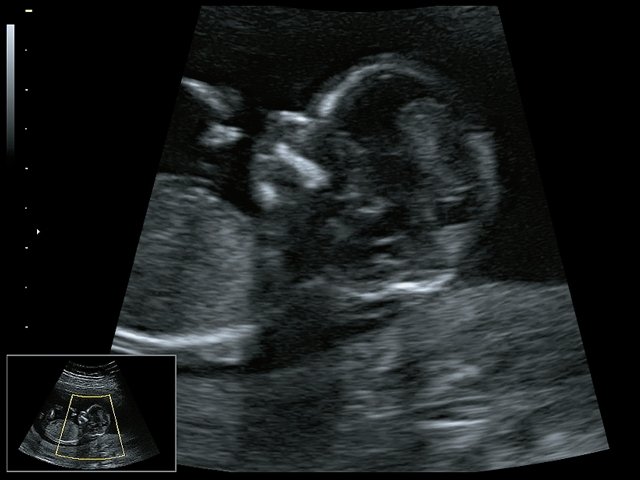
Рис. 2. Микрогнатия — сагиттальный скан в 2D, беременность 13 нед.
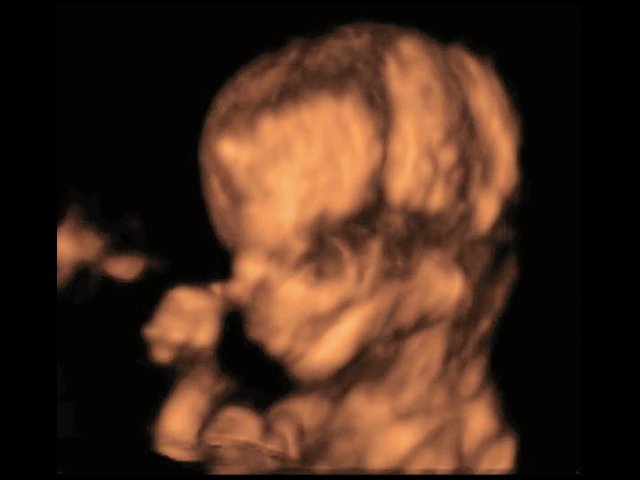
Рис. 3. Микрогнатия — 3D-реконструкция лица плода при СТК.
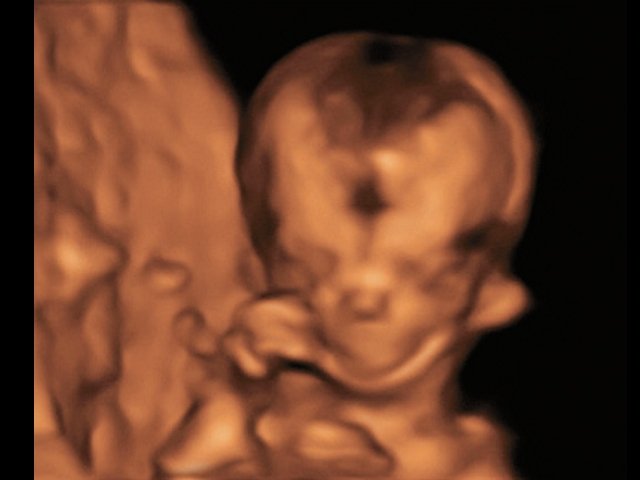
Рис. 4. Микрогнатия — 3D-реконструкция лица плода при синдроме Тричера Коллинза.
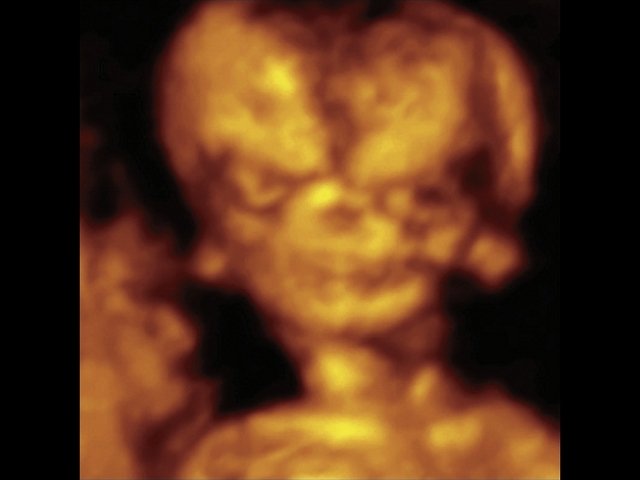
Рис. 5. Треугольная форма лица плода при синдроме Тричера Коллинза, аномальная форма и положение ушей в 3D.
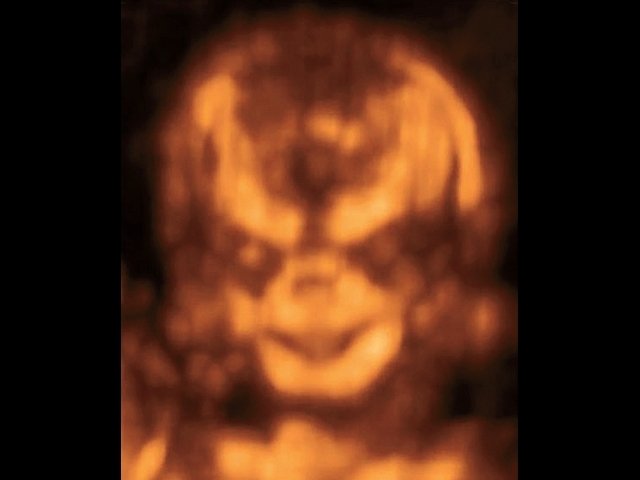
Рис. 6. 3D-сканирование лица плода с синдромом Тричера Коллинза. Опущенные книзу глазницы, гипоплазия скуловых костей.
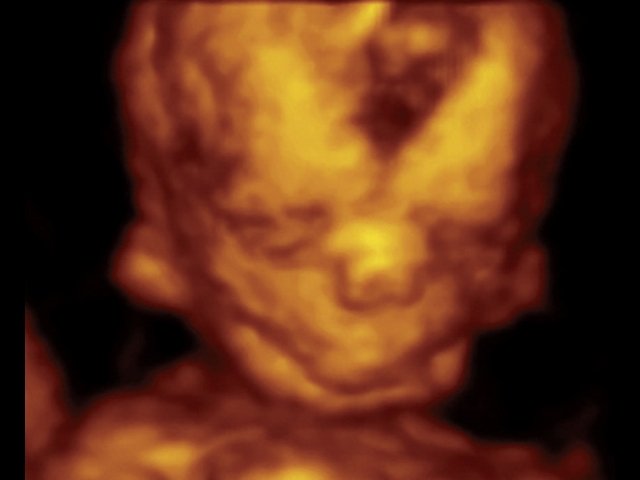
Рис. 7. Фенотип лица плода с синдромом Тричера Коллинза.
В МГНЦ по результату исследования ДНК плода в образцах плодного материала, полученного при проведении аспирации ворсин хориона, методом автоматического секвенирования у плода обнаружена патогенная мутация в гене TCOF1 (с. 3946- 3947 del GA) в гетерозиготном состоянии.
Выставлен клинический диагноз: беременность 13 нед. Синдром Тричера Коллинза у плода. Отягощенный генетический анамнез. Проведены медико-генетическое консультирование и пренатальный консилиум, прогноз для здоровья будущего ребенка определен как условно неблагоприятный. Семья приняла решение о досрочном прекращении беременности.
По результату повторного консультирования в МГО по вопросу репродуктивного поведения и прогноза потомства семья приняла решение о планировании следующей беременности с применением методик вспомогательных репродуктивных технологий с предимплантационной генетической диагностикой.
Выводы
- Оптимальным периодом для ультразвукового пренатального выявления новых случаев СТК (мутация de novo) является обследование во II триместре (в сроки второго скринингового обследования в 19–22 нед беременности), а для семейно-положительных случаев – период первого скринингового обследования (12–14 нед беременности).
- 3D/4D-изображения более наглядно и очевидно демонстрируют особенности изменения лицевого фенотипа при некоторых генетических синдромах, обеспечивая более понятную визуализацию признаков как для специалистов, так и для родителей.
- Сонографические находки при подозрении на СТК чрезвычайно важны, чтобы добавить специфическое обследование (секвенирование) в обычную панель исследования материала плодного происхождения, подтверждая диагноз.
- Учитывая варианты различной экспрессивности генов, необходимы тщательнейший осмотр пациентов (как матери, так и отца), сбор семейного анамнеза для постановки окончательного диагноза и определения риска повтора заболевания в семье и формулировки
специфических, конкретных мер профилактики генетической патологии.
Литература
- Кеннет Л. Джонс Наследственные синдромы по Д. Смиту М.: Практика, 2011: 296–297.
- https://omim.org/entry/154500?search=omim%20154500&highlight=154500%20omim
- Franceschetti A., Klein D. Mandibulo-facial dysostosis: new hereditary syndrome // Acta Ophthal. 1949; 27: 143–224.
- Bowman M., Oldridge M., Archer C. et al. Gross deletions in TCOF1 are a cause of Treacher-Collins-Franceschetti syndrome // Eur J Hum Genet. 2012; 20: 769–777.
- Edery P., Manach Y., Le Merrer M. et al. Apparent genetic homogeneity of the Treacher Collins-Franceschetti syndrome // Am J Med Genet. 1994; 52: 174–177.
- Trainor P.A., Dixon J., Dixon M.J. Treacher Collins syndrome: etiology, pathogenesis and prevention // Eur J Hum Genet. 2009; 17: 275–283.
- Beryl Benacerraf. Ultrasound of Fetal Syndromes. Elsevier, 2008: 184–186.
- Vincent M., Genevieve D., Ostertag A. et al. 44 others. Treacher Collins syndrome: a clinical and molecular study based on a large series of patients // Genet Med. 2016; 18: 49–56.
- Dixon M.J. Treacher Collins syndrome // Hum Mol Genet. 1996; 5 Spec No: 1391–1396.
- Teber O.A., Gillessen-Kaesbach G., Fischer S. et al. Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation // Eur J Hum Genet. 2004; 12: 879–890.
- Lowry R.B., Morgan K., Holmes T.M. et al. Mandibulofacial dysostosis in Hutterite sibs: a possible recessive trait // Am J Med Genet. 1985; 22: 501–512.
- Edwards S.J., Fowlie A., Cust M.P. et al. Prenatal diagnosis in Treacher Collins syndrome using combined linkage analysis and ultrasound imaging // J Med Genet. 1996; 33: 603–606.
- Ahmed, Ahmed Zakaria, Mahy Mohsen // https://sonoworld.com/TheFetus/page.aspx?id=3819 Treacher Collins syndrome.
- Nicolaides K.H., Johansson D., Donnani D., Rodeck C.H. Prenatal diagnosis of mandibulofacial dysostosis // Prenat Diagn. 1984; 4: 201–205.
- Cohen J., Ghezi F., Goncalves I. et al. Prenatal Sonographic diagnosis of Teacher Collins syndrome: a case and review of literature // Am J Perinatol. 1995; 12: 416–419.
- Tanaka Y, Kanenishi K., Tanaka H. et al. Antenatal three-dimensional sonographic features of Treacher Collins syndrome // Ultrasound Obstet Gynecol. 2002; 19: 414–415.
- Grochal F., Dankovcik R. et al. Treacher Collins syndrome – role of 3D/4D ultrasound in the assessment of fetal facial dysmorphism // https://sonoworld.com/TheFetus/page.aspx?id=3491
УЗИ аппарат HM70A
Экспертный класс по доступной цене. Монокристальные датчики, полноэкранный режим отображения, эластография, 3D/4D в корпусе ноутбука. Гибкая трансформация в стационарный сканер при наличии тележки.
Большая немедицинская проблема
Фильм «Чудо» режиссера Стивена Чбоски рассказывает историю мальчика Огги Пулмана с синдромом Тричера Коллинза. Мнения родителей, посмотревших кино вместе с детьми, разделились. Кто-то пишет в интернете восторженные отзывы: «Наконец-то в кинотеатрах идет трогательный и серьезный детский фильм без мата».
Кто-то сетует, что в экранизации обрезали подробности одноименной книги Ракель Паласио (2012), на основе которой создан фильм. «По сравнению с книжкой даже непонятно, почему главный герой – чудо. А ведь суть в том, что гены порой выстраиваются в редкие комбинации. И их сочетание может дать невероятную красоту или талант или, наоборот, болезнь, как это и случилось у героя. Но это не помешало ему быть настоящим чудом».
Специфического лечения синдрома Тричера Коллинза не существует, лечение заключается в комплексном устранении симптомов и их последствий (пластическая хирургия челюсти, ушных и иногда носовых проходов), мероприятия по восстановлению слуха и последующему развитию речи.
Пациенты с синдромом Тричера-Коллинза имеют сохранный интеллект, но могут иметь проблемы с речью, вызванные проблемами со слухом и, в особо тяжелых случаях, особенностями строения челюстей.
Главный герой фильма – сын богатых родителей, которые заботятся о нем, учат, а потом всей душой переживают, как их ребенок найдет общий язык с одноклассниками. Но в жизни – в российской практике – синдром Тричера Коллинза осложняется большой немедицинской проблемой: увидев, что родили ребенка-инвалида, мамы нередко оставляют таких детей в роддоме.
Кому-то кажется, что вырастить ребенка-инвалида – сложно и дорого. Кому-то неприятно осознавать, что в их генах есть проблемная мутация, и страшно иметь рядом «лицо» собственного заболевания.
В результате большая часть российских «детей без лица» – это сироты.
И поэтому к синдрому Тритчера Коллинза у наших героев, в отличие от киношного Огги, прибавился «синдром заброшенности» со всеми вытекающими.
Но нашлись две удивительные женщины, ставшие им мамами.
Максим, по меркам сиротской системы, невероятный везунчик
Как у многих людей с его диагнозом, у Максима – проблемы со слухом. Операции по реконструкции внутреннего уха делают только в Америке, это очень дорого. Поэтому единственный выход для Максима – слуховой аппарат.
Операцию на небе мальчику сделали еще в детдоме, в остальном он развивается нормально, а болеет даже реже своего младшего брата, кровного сына мамы Ани, взявшей Максима из детдома.
Аня узнала про Макса на форуме фонда «Волонтеры в помощь детям-сиротам»: много лет ему пытались найти семью, но никто не брал. И тогда Анна с мужем и сыновьями решились.
По меркам сиротской системы, Максим – невероятный везунчик – в свое время его распределили не в детский дом-интернат, где живут инвалиды, а в обыкновенный детский дом. Шансы попасть в семью из такого учреждения – гораздо выше, и это сработало.
Но еще до того, как познакомиться с Аней, Максим ездил на занятия в реабилитационный центр. Там его научили говорить – к счастью, успели до шести лет. Считается, если до шести лет у человека не появилась речь, этот навык будет потерян навсегда – дальше ребенок не заговорит, даже если начнет слышать.
На внешность Максима люди реагируют по-разному – кто-то показывает пальцем, кто-то не обращает внимания. Дети иногда говорят, что он «похож на зомби». Однажды девочка, увидев его в кинотеатре, заплакала.
В школе к ученику с необычной внешностью относятся спокойно. Правда, Максим пошел в школу для детей с проблемами по здоровью – там внешними особенностями не удивишь. В итоге слуховой аппарат Максима вызвал в классе даже больший интерес, чем он сам. Одноклассники спросили, что за странную штуку он носит на голове?
А вот на детской площадке как-то был казус: за Максимом по пятам стал бегать мальчик лет девяти и кричать: «Смотрите, он страшный!» Отстал только тогда, когда старшие ребята сказали: «Сам ты страшный. Прекрати орать, а то побьем». Максим в это время был без слухового аппарата, так что происходящего не расслышал.
А было и так: один мальчик посмотрел на него и сказал: «Может быть, его вылечить можно – операцию сделать? Надо, наверное, ему на операцию денег собрать».
В поликлинике, глядя на необычного пациента, гардеробщицы иногда удивляются: «Надо же, сам одевается!» Однажды, прямо в присутствии мамы, кто-то без спросу пытался натянуть на него куртку. Хотя Макс прекрасно одевается сам. Ну, а как не одеваться, – ребенку семь лет.
А еще Максим безумно любит всякую технику. Завести машину или переключить скорости для него – радость радостей. «Как только сын подрастет так, что ноги дотянутся до педалей, мы с папой, пожалуй, начнем прятать ключи от автомобиля», – смеется мама Аня.
Не так страшен синдром, как мифы о нем
Сам синдром Тричера-Коллинза врачи, как правило, неплохо опознают «в лицо», его сложно с чем-то спутать. Проблема в другом: в картах детдомовских детей поселяется ряд сопутствующих диагнозов, большинства из которых в семье было бы легко избежать.
Например, у Максима тугоухость привела к тому, что он начал говорить очень поздно – когда с помощью благотворителей попал на специальные занятия к логопеду.
Детдомовскому ребенку с задержкой речи в медицинской карте записывают диагноз «умственная отсталость», хотя физиологических оснований для этого нет.
К счастью, к тому времени, когда он познакомился со своей будущей мамой, Максим умел немного читать по слогам и писал печатными буквами. У шестилетнего ребенка с такими умениями страшные записи в медкарте сразу показались необоснованными.
«Я знаю пару, у которой ребенок с синдромом Тритчера Коллинза свой, – рассказывает Анна. – Ему слуховой аппарат надели в шесть месяцев, а не в шесть лет. В итоге мальчик нормально в три года пошел в сад.
Но в детском доме слуховой аппарат стоимостью в семьсот тысяч рублей – вещь невозможная. Дома-то в первый месяц приходилось следить не столько за Максом, сколько за аппаратом – чтобы не потерял и не разбил. В детском доме, где уронить или разломать вещь – обычное дело, такую дорогую технику даже не стремятся получить».
Когда Анна оформляла документы на ребенка, в опеке пугали: «вам этого ребенка не надо, возьмите другого», «вы видели список его диагнозов? – там расщелина неба, порок сердца и расщелина позвоночника». На самом деле расщелину неба Максиму прооперировали в два года, в сердце анализы показали небольшую особенность, которая считается вариантом нормы. Откуда взялось все остальное, а также особая «забота» опеки, – непонятно.
Сиротские последствия тяжелее всех синдромов
Руководитель программы «Близкие люди» фонда «Волонтеры в помощь детям-сиротам» Алена Синкевич: «Люди легко приспосабливаются к любому синдрому, но тяжело – к сиротским последствиям. Люди с генетическими синдромами рождались и будут рождаться. Но тяжелее всего они преодолевают последствия, возникшие во время, которые провели в детдоме одни».
Благотворительного фонда, который специально занимался бы проблемами людей с синдромом Тричера Коллинза, в России нет. Но скоро должен появиться фонд по решению проблем с кондуктивной тугоухостью, – у многих «людей без лица» есть такие проблемы. После этого жизнь детей с редким диагнозом станет чуть-чуть легче – разумеется, если о них позаботятся их семьи.
Макс долго не понимал, что люди растут
В жизни Максима есть проблемы и вовсе фантастические. Они никак не связаны с диагнозом, зато напрямую связаны с детдомовским прошлым. Макс долго не понимал, что люди растут.
Он знал, что есть маленькие люди – дети, – и есть взрослые, и думал, что навсегда останется маленьким.
А когда у Максима был первый день рождения дома, совсем не понял, с чем его поздравляют. Разговоры про «когда я вырасту» начались где-то через год жизни в семье.
А еще в детском доме с Максимом никто не говорил о маме и о том, как он здесь оказался. Даже о том, что Аня приехала его забирать, ребенку сказали в последний момент.
И некоторое время Максим был уверен, что Аня с мужем – его кровные родители. И что они когда-то отвезли его в детдом, а теперь вот забрали обратно.
И ему было совершенно непонятно, почему братьев – старшего и младшего – ни в какой детдом не отвозили.
Теперь Максим – почти обычный домашний ребенок. Ему нравится ходить в школу, правда, в магазин мама отпускает пока только вместе со старшим братом – семилетний Макс не очень хорошо считает.
Синдром Тричера-Коллинза или челюстно-лицевой дизостоз – наследственное заболевание, вызванное мутацией в гене TCOF1. Болезнь встречается у одного из 50 тысяч новорожденных. Проявляется недоразвитием лицевых костей черепа (часть их просто отсутствует).
Существует три степени проявленности заболевания. Люди с незначительными лицевыми дефектами могут не подозревать, что являются носителями генетической аномалии.
Челюстно-лицевой дизостоз средней степени приводит к возникновению характерной внешности с особой словно «утопленной» вглубь черепа формой лица, «грустными» глазами, несоразмерно маленьким ртом и подбородком. Часто люди с этой степенью заболевания имеют также недоразвитые ушные раковины и дефекты слуховых проходов, что приводит к проблемам со слухом и необходимости в слуховом аппарате. Могут встречаться разнообразные дефекты неба – от особого строения (так называемое высокое небо) до расщелин, которые корректируются с помощью пластических операций.
Сильная степень проявления челюстно-лицевого дизостоза – это грубые нарушения в строении лица, для коррекции которых бывает нужен не один десяток пластических операций.
Федя, мальчик с трубочками
У трехлетнего Феди синдром Тритчера Коллинза выражен больше, чем у Максима: он практически не слышит, не сформированы оба внутренних уха, есть проблемы с дыханием – непроходимость носовых ходов. Из-за этого в раннем детстве Федя не мог сосать. Обычный катетер через нос ему поставить не могли, в результате стали кормить через гастростому. А дышит Федя через трахеостому.
«Я иногда мечтаю, – говорит приемная мама Татьяна, – что с нас сняли стомы. Пусть даже синдром остается, а трубки бы сняли.
И Федя смог бы ходить в сад и школу, и не нужно будет постоянно возить с собой дезинфицирующие салфетки и литры мирамистина».
Татьяна уверена: если бы Федя жил в семье, и его нос вовремя бы прооперировали, одной трубки можно было избежать. У мальчика сформировался бы обычный навык еды, вкус. Пока же научить его есть обычную пищу не получается.
А для того, чтобы избавиться от проблем с дыханием и трахеостомы, Феде нужно пережить еще операцию по коррекции челюсти, возможно, не одну. Но и у мальчика с трубочками основные проблемы оказались не в них, а в тяжелых воспоминаниях.
Есть он боится
Для того чтобы в изоляторе детдома-интерната ребенок не сорвал с себя трубки и не задохнулся, его руки привязывали к кровати. В итоге Федя запомнил: трогать лицо нельзя, рот – запретная зона. Теперь поднести руки к лицу или есть ложкой он не может – боится. Обычную пищу Федя ест руками и почти всегда украдкой – вдруг заругают.
«Первое впечатление от Феди у меня было шоковое, – рассказывает Татьяна, ходившая в детдом волонтерить. – И с синдромом это было связано меньше всего.
Гораздо страшнее отсутствия ушек был рост полуторогодовалого Феди – всего 60 сантиметров. Такой рост никак не связан с его диагнозом, это результат депривации.
К тому времени большую часть своей жизни Федя находился в изоляторе и лежал. От лежания его затылок сплющился.
На фоне недостаточно выдвинутой нижней челюсти лоб казался нависающим и непропорционально огромным. На первый взгляд, это был не ребенок, а зародыш».
Постоянно лежащему глазами в потолок ребенку было почти не на что смотреть – это привело к проблемам со зрением. Они также не связаны с Фединым синдромом, это – последствия времени, проведенного в одиночестве. С непривычки Федя с трудом фокусировал взгляд на предметах и довольно сильно косил. Сейчас, после двух лет дома, эта особенность почти исчезла.
А еще мальчик никогда не бывал на солнце, и поэтому был синего цвета. Удивительно, как в этих условиях вообще захотел выжить.
Как и от Максима, мама отказалась от Феди в роддоме сразу после рождения. Татьяна решила взять Федю, потому что понимала: в детдоме ребенок погибнет, а вот в семье ему можно будет помочь. Сотрудники опеки брать Федю отговаривали: «А давайте мы вам кого-нибудь еще покажем. У нас большой банк данных».
Окружающие – друзья и знакомые семьи – на Федю реагируют спокойно. Ну а дети при первой встрече могут спросить: «А что с ним … не так?»
Правда, помимо необычной внешности, у мальчика есть еще ряд особенностей.
С раннего детства Федя почти не видел людей, общаться и разговаривать не привык. Поэтому родителей в первые полтора года дома «не замечал» – в детдоме не понял, что от людей бывает польза.
Сейчас Федя старается изучать мир вокруг: например, если оказывается в незнакомом помещении, он не пугается и не бежит к маме, а идет и внимательно осматривает новые предметы.
Вещи, в отличие от людей, вызывают у Феди живейший интерес.
Впрочем, речь в последние полгода у Феди тоже появилась. Татьяна надеется, что дело пойдет значительно быстрее, когда Феде поставят слуховой имплант, и слух будет постоянным.
Татьяна: «Если бы у нас маму, родившую ребенка с какой-то внешней патологией, не пугали, а сразу говорили: «Это решается. В два года сделаешь ему такую операцию, в три – следующую, потом будете заниматься слухом, развивать речь. Вот это покрывается страховкой, с этой болезнью работают такие благотворительные фонды, а вот здесь находится сообщество мамочек детей с таким же диагнозом», – то есть предлагали бы пошаговую маршрутизацию в решении ее проблемы – у людей не было бы такого страха, и детей не оставляли бы в детдомах».
Вы можете помочь!
Федя и другие дети с редкими синдромами находятся под опекой Детского хосписа «Дом с маяком». Вы можете помочь, отправив смс со словом «Дети» на номер 1200 с любой суммой. Например: Дети 500. Не забудьте ответить на смс-подтверждение, иначе ваша помощь не дойдет.
Другие способы перевода:
Пожалуйста, указывайте в назначении платежа «Дети»
– С карты:
– PayPal:
– Завезти наличные в кассу Детского хосписа на м.Преображенская площадь, в рабочее время.
– Перевести на счет Детского хосписа. Банковские реквизиты:
Благотворительное медицинское частное учреждение «Детский хоспис»
ИНН/КПП 7704280903/770401001
р/с № 40703810238180000837
в ПАО «Сбербанк России» г.Москва
БИК 044525225
к/с 30101810400000000225
Сайт «Милосердие» благодарит Детский хоспис «Дом с маяком» и проект «Близкие люди» фонда «Волонтеры в помощь детям-сиротам» за содействие в подготовке материала.
Существует много генетических заболеваний, приводящих к тяжелым аномалиям и мутациям. Одной из таких патологий является синдром Тричера Коллинза. Это тяжелое заболевание, приводящее к обезображиванию лица. Обнаруживается патология на начальном этапе внутриутробного развития или же после рождения ребенка. Примерно 1 новорожденный из 50 тысяч поражен этой загадочной болезнью. Патология пока плохо изучена, хотя сейчас в мире проживает довольно много людей с различными стадиями болезни.
Что такое синдром Тричера Коллинза
Патология была названа так в честь английского офтальмолога, впервые описавшего ее в начале 20 века. По-другому болезнь называется челюстно-лицевой дизостоз. Она представляет собой генетическое врожденное заболевание, имеющее доминантный тип наследования, то есть в 100% передающееся по наследству.
Синдром Тричера Коллинза – это отклонение в развитии, выражающееся в дефектах развития костей черепа. При этом оказывается обезображено лицо. Недоразвитие костей челюсти, скуловых, лобных и других приводит к появлению характерных уродств. Заболевание врожденное, поэтому аномалии видны сразу при рождении ребенка, если не удалось их выявить во внутриутробном развитии.
В большинстве случаев патология не опасна для жизни, а также не влияет на умственное и физическое развитие ребенка. Но иногда деформации черепа настолько серьезны, что ребенок не может самостоятельно дышать и есть.
Почему появляется заболевание
Синдром Тричера Коллинза развивается в результате генетической мутации. Она появляется в группе генов, отвечающих за построение тканей в районе черепа и лица. Именно с помощью этих генов на раннем этапе внутриутробного развития происходит формирование лица ребенка. Но иногда случается сбой в их работе. До сих пор учеными не выяснены причины такой патологии. Примерно в половине случаев заболевание наследуется от одного или обоих родителей. Этот признак является доминантным, поэтому в семье с этой патологией обязательно родятся больные дети.
Но почти 60% детей получают болезнь от здоровых родителей. Сложно сказать, почему это происходит. Многие факторы могут влиять на гены ребенка. А ген, ответственный за построение костей черепа, может повреждаться по таким причинам:
- при употреблении женщиной алкоголя, наркотиков или курения на раннем этапе беременности;
- при серьезных стрессовых ситуациях;
- из-за поражения женщины инфекциями, например, токсоплазмозом или цитомегаловирусом;
- вследствие тяжелых хронических заболеваний;
- при приеме некоторых лекарственных препаратов.

Синдром Тричера Коллинза – это генетическое заболевание, передающееся по наследству
Симптомы
У каждого человека заболевание проявляется по-разному. Могут наблюдаться один или два симптома, или же полный набор признаков. Но ребенка с синдромом Тричера Коллинза можно узнать сразу. Фото больных детей есть в интернете, часто они похожи друг на друга. У заболевания могут быть такие внешние признаки:
- широкий разрез глаз, недоразвитие нижнего века, верхнее веко может приобретать треугольную форму, а внешний край глаза направлен вниз;
- нос по сравнению с другими костями лица очень крупный;
- из-за недоразвития скуловых костей лицо кажется маленьким, «птичьим»;
- очень маленький подбородок, нижняя челюсть иногда отсутствует, поэтому часто язык не вмещается в ротовую полость;
- «волчья пасть» – отсутствие костей верхнего неба;
- «заячья губа» – расщепление кости верхней челюсти;
- недоразвитие слуховых костей, отсутствие ушных раковин;
- рост волос на щеках;
- деформация больших пальцев на руках.
Стадии развития болезни
Признаки синдрома Тричера Коллинза могут проявляться, начиная от почти незаметных до очень тяжелых деформаций. В соответствии с этим выделяют три стадии развития заболевания.
- На начальной стадии деформации костей практически незаметны. Дети с такой патологией могут вести нормальную жизнь.
- Вторая стадия характеризуется серьезными аномалиями развития костей черепа. К ним присоединяются патологии слуха, зубов, сложности с дыханием или принятием пищи.
- Третья самая тяжелая стадия заболевания характеризуется почти полным отсутствием лица у ребенка.

В легких случаях патология почти не сильно заметна внешне
Лечение
Часто аномалию выявляют в период внутриутробного развития. Маме рекомендуют прервать беременность. Если же этого не произошло, ребенку предстоит сложное и длительное лечение. Но особенность его в том, что исправить деформацию можно только с помощью операции. Часто необходимо проведение множества пластических операций. Но не всегда удается устранить все аномалии. Если же реконструкция костей лица прошла успешно, дети после синдрома Тричера Коллинза могут вести нормальную жизнь.
Кроме операций по исправлению костных деформаций, таким больным часто требуется слухопротезирование. Ведь аномалии в большинстве случаев распространяются на слуховой проход. Потому многие больные страдают тугоухостью. А при сильной деформации челюстных костей необходимы стоматологические операции по восстановлению зубов и исправлению прикуса. Иногда больным требуется трахеотомия, так как в тяжелых случаях они не могут дышать самостоятельно.
Осложнения
Синдром Тричера Коллинза опасен своими последствиями. Чаще всего – это тугоухость. Ведь заболевание характеризуется недоразвитием слуховых костей, часто также отсутствуют ушные раковины. Серьезным последствием является недоразвитие ротового аппарата. Деформация челюстей и зубов, отсутствие слюнных желез – это приводит к невозможности самостоятельно принимать пищу. Может быть также нарушена функция дыхания из-за зарастания носовых ходов, недоразвития верхнего неба и большого размера языка, который перекрывает дыхательные пути.
Иногда эта генетическая патология приводит к аномалиям развития других органов, например, к пороку сердца. Часто развивается косоглазие, стоматологические заболевания.
Как живут дети с синдромом
Болезнь очень часто не проявляется больше никакими признаками, кроме внешних деформаций лица. Поэтому больные с синдромом Тричера Коллинза могут успешно адаптироваться в обществе и развиваться в соответствии с возрастными нормами. Иногда им требуется логопедическая помощь, чтобы исправить нарушения речи из-за патологий челюсти.
Некоторые пациенты проходят длительную психологическую реабилитацию, так как видят свое уродство и страдают из-за этого. Поэтому иногда такие дети впадают в депрессию, испытывают трудности с общением.
Синдром Тричера Коллинза – это пожизненный диагноз. Но многие больные смирились со своей патологией и ведут нормальную жизнь.
| Treacher Collins syndrome | |
|---|---|
| Other names | Treacher Collins–Franceschetti syndrome,[1] mandibulofacial dysostosis,[2] Franceschetti-Zwalen-Klein syndrome[3] |
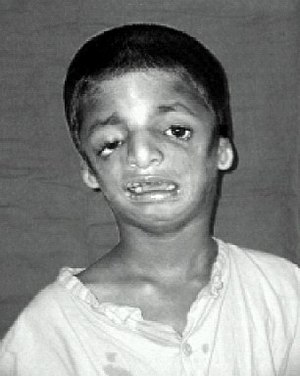 |
|
| Child with Treacher Collins syndrome[4] | |
| Specialty | Medical genetics |
| Symptoms | Deformities of the ears, eyes, cheekbones, chin[5] |
| Complications | Breathing problems, problems seeing, hearing loss[5] |
| Causes | Genetic[5] |
| Diagnostic method | Based on symptoms, X-rays, genetic testing[3] |
| Differential diagnosis | Nager syndrome, Miller syndrome, hemifacial microsomia[3] |
| Treatment | Reconstructive surgery, hearing aids, speech therapy[6] |
| Prognosis | Generally normal life expectancy[6] |
| Frequency | 1 in 50,000 people[5] |
Treacher Collins syndrome (TCS) is a genetic disorder characterized by deformities of the ears, eyes, cheekbones, and chin.[5] The degree to which a person is affected, however, may vary from mild to severe.[5] Complications may include breathing problems, problems seeing, cleft palate, and hearing loss.[5] Those affected generally have normal intelligence.[5]
TCS is usually autosomal dominant.[5] More than half the time it occurs as a result of a new mutation rather than being inherited.[5] The involved genes may include TCOF1, POLR1C, or POLR1D.[5] Diagnosis is generally suspected based on symptoms and X-rays, and potentially confirmation by genetic testing.[3]
Treacher Collins syndrome is not curable.[6] Symptoms may be managed with reconstructive surgery, hearing aids, speech therapy, and other assistive devices.[6] Life expectancy is generally normal.[6] TCS occurs in about one in 50,000 people.[5] The syndrome is named after Edward Treacher Collins, an English surgeon and ophthalmologist, who described its essential traits in 1900.[7][8]
Signs and symptoms[edit]

The same child shown from the front above in infobox, now seen from the side, with small ears and a chin that is far back.[4]
Symptoms in people with Treacher Collins syndrome vary. Some individuals are so mildly affected that they remain undiagnosed, while others have moderate to severe facial involvement and life-threatening airway compromise.[9] Most of the features of TCS are symmetrical and are already recognizable at birth.[citation needed]
The most common symptom of Treacher Collins syndrome is underdevelopment of the lower jaw and underdevelopment of the zygomatic bone. This can be accompanied by the tongue being retracted. The small mandible can result in a poor occlusion of the teeth or in more severe cases, trouble breathing or swallowing. The respiratory system of a child with Treacher Collins syndrome is the primary concern at birth, with other issues only addressed once respiratory function has been stabilized.[10] Underdevelopment of the zygomatic bone gives the cheeks a sunken appearance.[11][12]
The external ear is sometimes small, rotated, malformed, or absent entirely in people with TCS. Symmetric, bilateral narrowing or absence of the external ear canal, is also described.[12][13] In most cases, the bones of the middle ear and the middle ear cavity are misshapen. Inner ear malformations are rarely described. As a result of these abnormalities, a majority of the individuals with TCS have conductive hearing loss.[12][14]
Most affected people also experience eye problems, including coloboma (notches) in the lower eyelids, partial or complete absence of eyelashes on the lower lid, downward angled eyelids, drooping of upper and lower eyelids, and narrowing of the tear ducts. Vision loss can occur and is associated with strabismus, refractive errors, and anisometropia. It can also be caused by severely dry eyes, a consequence of lower eyelid abnormalities and frequent eye infections.[12][13][15][16]
Although an abnormally shaped skull is not distinctive for Treacher Collins syndrome, brachycephaly with bitemporal narrowing is sometimes observed.[13] Cleft palate is also common.[12]
Dental anomalies are seen in 60% of affected people, including tooth agenesis (33%), discoloration (enamel opacities) (20%), malplacement of the maxillary first molars (13%), and wide spacing of the teeth. In some cases, dental anomalies in combination with mandible hypoplasia result in a malocclusion. This can lead to problems with food intake and the ability to close the mouth.[12]
Less common features of TCS may add to an affected person’s breathing problems, including sleep apnea. Choanal atresia or stenosis is a narrowing or absence of the choanae, the internal opening of the nasal passages, which may also be observed. Underdevelopment of the pharynx can also narrow the airway.[12]
Features related to TCS that are seen less frequently include nasal deformities, high-arched palate, macrostomia, preauricular hair displacement, cleft palate, hypertelorism, notched upper eyelid, and congenital heart defects.[11][12][16]
Although facial deformity is often associated with developmental delay and intellectual disability, more than 95% of people affected with TCS have normal intelligence.[12] The psychological and social problems associated with facial deformity can affect quality of life in individuals with TCS.
Genetics[edit]

Mutations in TCOF1, POLR1C, or POLR1D genes can cause Treacher Collins syndrome.[17] TCOF1 gene mutations are the most common cause of the disorder, with POLR1C and POLR1D gene mutations causing an additional 2% of cases. In individuals without an identified mutation in one of these genes, the genetic cause of the condition is unknown. The TCOF1, POLR1C, and POLR1D genes code for proteins which play important roles in the early development of bones and other tissues of the face. Mutations in these genes reduce the production of rRNA, which may trigger the self-destruction (apoptosis) of certain cells involved in the development of facial bones and tissues. It is unclear why the effects of a reduction in rRNA are limited to facial development. Mutations in TCOF1 and POLR1D cause the autosomal dominant form of Treacher Collins, and mutations in POLR1C cause the autosomal recessive form.[12]
TCOF1[edit]
TCOF1 is the primary gene associated with TCS, a mutation in this gene being found in 90–95% of the individuals with TCS.[11][18] However, in some individuals with typical symptoms of TCS, mutations in TCOF1 have not been found.[19] Investigation of the DNA has resulted in the identification of the kind of mutations found in TCOF1. The majority of mutations are small deletions or insertions, though splice site and missense mutations also have been identified.[11][20][21][22]
Mutation analysis has unveiled more than 100 disease-causing mutations in TCOF1, which are mostly family-specific mutations. The only recurrent mutation accounts for about 17% of the cases.[23]
TCOF1 is found on the 5th chromosome in the 5q32 region. It codes for a relatively simple nucleolar protein called treacle, that is thought to be involved in ribosome assembly.[18] Mutations in TCOF1 lead to haploinsufficiency of the treacle protein.[24] Haploinsufficiency occurs when a diploid organism has only one functional copy of a gene, because the other copy is inactivated by a mutation. The one normal copy of the gene does not produce enough protein, causing disease. Haploinsufficiency of the treacle protein leads to a depletion of the neural crest cell precursor, which leads to a reduced number of crest cells migrating to the first and second pharyngeal arches. These cells play an important role in the development of the craniofacial appearance, and loss of one copy of treacle affects the cells’ ability to form the bones and tissues of the face.[12][20][25]
Other mutations[edit]
POLR1C and POLR1D mutations are responsible for a minority of cases of Treacher Collins. POLR1C is found on chromosome 6 at position 6q21.2 and POLR1D is found on chromosome 13 at position 13q12.2. Those genes code for a protein subunits shared between RNA polymerase I and III. Both of these polymerases are important for ribosome biogenesis.[12]
Diagnosis[edit]
Genetic counseling[edit]
TCS is inherited in an autosomal dominant manner and the penetrance of the affected gene is almost complete.[26] Some recent investigations, though, described some rare cases in which the penetrance in TCS was not complete. Causes may be a variable expressivity, an incomplete penetrance[27] or germline mosaicism.[28] Only 40% of the mutations are inherited. The remaining 60% are a result of a de novo mutation, where a child has a new mutation in the responsible gene and did not inherit it from either parent.[12][29] In the outcome of the disease, inter- and intrafamilial variability occurs. This suggests that when an affected child is born, it is important to investigate the parents to determine whether the affected gene is present, because the parent could have a mild form of the disease that has not been diagnosed. In this case, the risk of having another affected child is 50%. If the parents do not have the affected gene, the recurrence risk appears to be low.[26] In following generations, the severity of the clinical symptoms increases.[21]
Prenatal diagnosis[edit]
Mutations in the main genes responsible for TCS can be detected with chorionic villus sampling or amniocentesis. Rare mutations may not be detected by these methods. Ultrasonography can be used to detect craniofacial abnormalities later in pregnancy, but may not detect milder cases.[12]
Clinical findings[edit]
TCS is often first suspected with characteristic symptoms observed during a physical exam. However, the clinical presentation of TCS can resemble other diseases, making diagnosis difficult.[30] The OMENS classification was developed as a comprehensive and stage-based approach to differentiate the diseases. This acronym describes five distinct dysmorphic manifestations, namely orbital asymmetry, mandibular hypoplasia, auricular deformity, nerve development, and soft-tissue disease.[31]
Orbital symmetry
- O0: normal orbital size, position
- O1: abnormal orbital size
- O2: abnormal orbital position
- O3: abnormal orbital size and position
Mandible
- M0: normal mandible
- M1: small mandible and glenoid fossa with short ramus
- M2: ramus short and abnormally shaped
- 2A: glenoid fossa in anatomical acceptable position
- 2B: Temperomandibular joint inferiorly (TMJ), medially, anteriorly displaced, with severely hypoplastic condyle
- M3: Complete absence of ramus, glenoid fossa, and TMJ
Ear
- E0: normal ear
- E1: Minor hypoplasia and cupping with all structures present
- E2: Absence of external auditory canal with variable hypoplasia of the auricle
- E3: Malposition of the lobule with absent auricle, lobular remnant usually inferior anteriorly displaced
Facial nerve
- N0: No facial nerve involvement
- N1: Upper facial nerve involvement (temporal or zygomatic branches)
- N2: Lower facial nerve involvement (buccal, mandibular or cervical)
- N3: All branches affected
Soft tissue
- S0: No soft tissue or muscle deficiency
- S1: Minimal tissue or muscle deficiency
- S2: Moderate tissue or muscle deficiency
- S3: Severe tissue or muscle deficiency
Radiographs[edit]
A few techniques are used to confirm the diagnosis in TCS.[30][32]
An orthopantomogram (OPG) is a panoramic dental X-ray of the upper and lower jaw. It shows a two-dimensional image from ear to ear. Particularly, OPG facilitates an accurate postoperative follow-up and monitoring of bone growth under a mono- or double-distractor treatment. Thereby, some TCS features could be seen on OPG, but better techniques are used to include the whole spectrum of TCS abnormalities instead of showing only the jaw abnormalities.[30]
Another method of radiographic evaluation is taking an X-ray image of the whole head. The lateral cephalometric radiograph in TCS shows hypoplasia of the facial bones, like the malar bone, mandible, and the mastoid.[30]
Finally, occipitomental radiographs are used to detect hypoplasia or discontinuity of the zygomatic arch.[32]
CT scan[edit]
A temporal-bone CT using thin slices makes it possible to diagnose the degree of stenosis and atresia of the external auditory canal, the status of the middle ear cavity, the absent or dysplastic and rudimentary ossicles, or inner ear abnormalities such as a deficient cochlea. Two- and three-dimensional CT reconstructions with VRT and bone and skin-surfacing are helpful for more accurate staging and the three-dimensional planning of mandibular and external ear reconstructive surgery.[citation needed]
Differential diagnosis[edit]
Other diseases have similar characteristics to Treacher Collins syndrome. In the differential diagnosis, one should consider the acrofacial dysostoses. The facial appearance resembles that of Treacher Collins syndrome, but additional limb abnormalities occur in those persons. Examples of these diseases are Nager syndrome and Miller syndrome.[citation needed]
The oculoauriculovertebral spectrum should also be considered in the differential diagnosis. An example is hemifacial microsomia, which primarily affects development of the ear, mouth, and mandible. This anomaly may occur bilaterally. Another disease which belongs to this spectrum is Goldenhar syndrome, which includes vertebral abnormalities, epibulbar dermoids and facial deformities.[33]
Treatment[edit]
The treatment of individuals with TCS may involve the intervention of professionals from multiple disciplines. The primary concerns are breathing and feeding, as a consequence of the hypoplasia of the mandibula and the obstruction of the hypopharynx by the tongue. Sometimes, they may require a tracheostomy to maintain an adequate airway,[34] and a gastrostomy to assure an adequate caloric intake while protecting the airway. Corrective surgery of the face is performed at defined ages, depending on the developmental state.[35]
An overview of the present guidelines:
- If a cleft palate is present, the repair normally takes place at 9–12 months old. Before surgery, a polysomnography with a palatal plate in place is needed. This may predict the postoperative situation and gives insight on the chance of the presence of sleep apnea (OSAS) after the operation.[11][36][37]
- Hearing loss is treated by bone conduction amplification, speech therapy, and educational intervention to avoid language/speech problems. The bone-anchored hearing aid is an alternative for individuals with ear anomalies.[38]
- Zygomatic and orbital reconstruction is performed when the cranio-orbitozygomatic bone is completely developed, usually at the age of 5–7 years. In children, an autologous bone graft is mostly used. In combination with this transplantation, lipofilling can be used in the periorbital area to get an optimal result of the reconstruction.[citation needed] Reconstruction of the lower eyelid coloboma includes the use of a myocutaneous flap, which is elevated and in this manner closes the eyelid defect.[39]
- External ear reconstruction is usually done when the individual is at least eight years old. Sometimes, the external auditory canal or middle ear can also be treated.
- The optimal age for the maxillomandibular reconstruction is controversial; as of 2004, this classification has been used:[11]
- Type I (mild) and Type IIa (moderate) 13–16 years
- Type IIb (moderate to severe malformation) at skeletal maturity
- Type III (severe) 6–10 years
- When the teeth are cutting, the teeth should be under supervision of an orthodontist to make sure no abnormalities occur. If abnormalities like dislocation or an overgrowth of teeth are seen, appropriate action can be undertaken as soon as possible.[20]
- Orthognathic treatments usually take place after the age of 16 years; at this point, all teeth are in place and the jaw and dentition are mature. Whenever OSAS is detected, the level of obstruction is determined through endoscopy of the upper airways. Mandibular advancement can be an effective way to improve both breathing and æsthetics, while a genioplasty only restores the profile.[11]
- If a nose reconstruction is necessary, it is usually performed after the orthognathic surgery and after the age of 18 years.[11]
- The contour of the facial soft tissues generally requires correction at a later age, because of the facial skeletal maturity. The use of microsurgical methods, like the free flap transfer, has improved the correction of facial soft tissue contours.[40] Another technique to improve the facial soft tissue contours is lipofilling. For instance, lipofilling is used to reconstruct the eyelids.[39]
Hearing loss[edit]
Hearing loss in Treacher Collins syndrome is caused by deformed structures in the outer and middle ear. The hearing loss is generally bilateral with a conductive loss of about 50–70 dB. Even in cases with normal auricles and open external auditory canals, the ossicular chain is often malformed.[41]
Attempts to surgically reconstruct the external auditory canal and improve hearing in children with TCS have not yielded positive results.[42]
Auditory rehabilitation with bone-anchored hearing aids (BAHAs) or a conventional bone conduction aid has proven preferable to surgical reconstruction.[38]
Psychiatric[edit]
The disorder can be associated with a number of psychological symptoms, including anxiety, depression, social phobia, and distress about body image. People who have this disorder may also experience discrimination, bullying, and name calling, especially when young. A multi-disciplinary team and parental support should include these issues.[43]
Epidemiology[edit]
TCS occurs in about one in 50,000 births in Europe.[44] Worldwide, it is estimated to occur in one in 10,000 to one in 50,000 births.[12]
History[edit]
The syndrome is named after Edward Treacher Collins (1862–1932), the English surgeon and ophthalmologist who described its essential traits in 1900.[7][8][45] In 1949, Adolphe Franceschetti and David Klein described the same condition on their own observations as mandibulofacial dysostosis. The term mandibulofacial dysostosis is used to describe the clinical features.[46]
Culture[edit]
In July 1977, a New York Times article describing new plastic surgery techniques which could partially correct the appearance of those with Treacher Collins syndrome was widely circulated resulting in raised awareness of the disease.[47]
Prior to beginning his comedy career, Bob Saget made a documentary short called «Through Adam’s Eyes» documenting his young nephew’s experiences undergoing facial reconstructive surgery due to Treacher Collins; the film won a student Academy Award.[48]
The disorder was featured on the show Nip/Tuck, in the episode «Blu Mondae».[49] TLC’s Born Without a Face[50] features Juliana Wetmore, who was born with the most severe case in medical history of this syndrome and is missing 30%–40% of the bones in her face.[50]
In 2010, BBC Three documentary Love Me, Love My Face[51] covered the case of a man, Jono Lancaster, with the condition. In 2011, BBC Three returned to Jono to cover his and his partner Laura’s quest to start a family,[2] in So What If My Baby Is Born Like Me?,[52] which first aired as part of a BBC Three season of programmes on parenting.[53] The first film was replayed on BBC One shortly ahead of the second film’s initial BBC Three broadcast. Lancaster’s third BBC Three film, Finding My Family on Facebook, which looked at adoption, aired in 2011.[54]
In Wonder, a children’s novel, the main character is a child who has Treacher Collins syndrome.[55] A 2017 film adaptation of Wonder, starring Julia Roberts, Owen Wilson and Jacob Tremblay, was released in November 2017.[56][57]
Alison Midstokke, who appears in the drama film Happy Face (2018),[58] is an actress and activist who has the condition.
See also[edit]
- First arch syndrome
- Franceschetti-Klein syndrome
- Hearing loss with craniofacial syndromes
References[edit]
- ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. pp. 894, 1686. ISBN 978-1-4160-2999-1.
- ^ a b «I hated seeing my face in the mirror». BBC Online. 18 November 2010. Archived from the original on 2018-10-11. Retrieved 2010-11-18.
- ^ a b c d «Treacher Collins Syndrome». NORD (National Organization for Rare Disorders). 2016. Archived from the original on 20 December 2019. Retrieved 7 November 2017.
- ^ a b Goel, L; Bennur, SK; Jambhale, S (August 2009). «Treacher-collins syndrome-a challenge for anaesthesiologists». Indian Journal of Anaesthesia. 53 (4): 496–500. PMC 2894488. PMID 20640217.
- ^ a b c d e f g h i j k l «Treacher Collins syndrome». Genetics Home Reference. June 2012. Archived from the original on 26 June 2020. Retrieved 7 November 2017.
- ^ a b c d e «Treacher Collins syndrome». rarediseases.info.nih.gov. 2015. Archived from the original on 16 August 2019. Retrieved 7 November 2017.
- ^ a b R, Pramod John; John, Pramod (2014). Textbook of Oral Medicine. JP Medical Ltd. p. 76. ISBN 9789350908501. Archived from the original on 2020-09-12. Retrieved 2017-09-17.
- ^ a b Beighton, Greta (2012). The Man Behind the Syndrome. Springer Science & Business Media. p. 173. ISBN 9781447114154.
- ^ Edwards, S J; Fowlie, A; Cust, M P; Liu, D T; Young, I D; Dixon, M J (1 July 1996). «Prenatal diagnosis in Treacher Collins syndrome using combined linkage analysis and ultrasound imaging». Journal of Medical Genetics. 33 (7): 603–606. doi:10.1136/jmg.33.7.603. PMC 1050672. PMID 8818950.
- ^ Mouthon, L., Busa, T., Bretelle, F., Karmous‐Benailly, H., Missirian, C., Philip, N., & Sigaudy, S. (2019). Prenatal diagnosis of micrognathia in 41 fetuses: Retrospective analysis of outcome and genetic etiologies. American Journal of Medical Genetics Part A, 179(12), 2365–2373. doi: 10.1002/ajmg.a.61359
- ^ a b c d e f g h Katsanis SH, et al., Treacher Collins syndrome, 2004, GeneReviews
- ^ a b c d e f g h i j k l m n o «The Physician’s Guide to Treacher Collins Syndrome» (PDF). National Organization for Rare Disorders (NORD). 2012. Archived from the original (PDF) on 2017-01-28.
- ^ a b c Posnick, Jeffrey C (1 October 1997). «Treacher Collins syndrome: Perspectives in evaluation and treatment». Journal of Oral and Maxillofacial Surgery. 55 (10): 1120–1133. doi:10.1016/S0278-2391(97)90294-9. PMID 9331237.
- ^ Trainor, Paul A; Dixon, Jill; Dixon, Michael J (24 December 2008). «Treacher Collins syndrome: etiology, pathogenesis and prevention». European Journal of Human Genetics. 17 (3): 275–283. doi:10.1038/ejhg.2008.221. PMC 2986179. PMID 19107148.
- ^ Hertle, R W; Ziylan, S; Katowitz, J A (1 October 1993). «Ophthalmic features and visual prognosis in the Treacher-Collins syndrome». British Journal of Ophthalmology. 77 (10): 642–645. doi:10.1136/bjo.77.10.642. PMC 504607. PMID 8218033.
- ^ a b Marszałek, B; Wójcicki, P; Kobus, K; Trzeciak, WH (2002). «Clinical features, treatment and genetic background of Treacher Collins syndrome». Journal of Applied Genetics. 43 (2): 223–33. PMID 12080178.
- ^ «Treacher Collins Syndrome». NORD (National Organization for Rare Disorders). Archived from the original on 2019-12-20. Retrieved 2016-02-29.
- ^ a b Dixon, Jill; Edwards, Sara J.; Gladwin, Amanda J.; Dixon, Michael J.; Loftus, Stacie K.; Bonner, Cynthia A.; Koprivnikar, Kathryn; Wasmuth, John J. (31 January 1996). «Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome». Nature Genetics. 12 (2): 130–136. doi:10.1038/ng0296-130. PMID 8563749. S2CID 34312227.
- ^ Teber OA, Gillessen-Kaesbach G, Fischer S, Böhringer S, Albrecht B, Albert A, Arslan-Kirchner M, Haan E, Hagedorn-Greiwe M, Hammans C, Henn W, Hinkel GK, König R, Kunstmann E, Kunze J, Neumann LM, Prott EC, Rauch A, Rott HD, Seidel H, Spranger S, Sprengel M, Zoll B, Lohmann DR, Wieczorek D (2004). «Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation». European Journal of Human Genetics. 12 (11): 879–90. doi:10.1038/sj.ejhg.5201260. PMID 15340364.
- ^ a b c Dixon, J; Trainor, P.; Dixon, M. J. (1 May 2007). «Treacher Collins syndrome». Orthodontics & Craniofacial Research. 10 (2): 88–95. doi:10.1111/j.1601-6343.2007.00388.x. PMID 17552945.
- ^ a b Masotti, Cibele; Ornelas, Camila C.; Splendore-Gordonos, Alessandra; Moura, Ricardo; Félix, Têmis M.; Alonso, Nivaldo; Camargo, Anamaria A.; Passos-Bueno, Maria (1 January 2009). «Reduced transcription of TCOF1 in adult cells of Treacher Collins syndrome patients». BMC Medical Genetics. 10 (1): 136. doi:10.1186/1471-2350-10-136. PMC 2801500. PMID 20003452.
- ^ Sakai, Daisuke; Trainor, Paul A. (31 May 2009). «Treacher Collins syndrome: Unmasking the role of Tcof1/treacle». The International Journal of Biochemistry & Cell Biology. 41 (6): 1229–1232. doi:10.1016/j.biocel.2008.10.026. PMC 3093759. PMID 19027870.
- ^ Splendore, Alessandra; Fanganiello, Roberto D.; Masotti, Cibele; Morganti, Lucas S. C.; Rita Passos-Bueno, M. (1 May 2005). «TCOF1 mutation database: Novel mutation in the alternatively spliced exon 6A and update in mutation nomenclature». Human Mutation. 25 (5): 429–434. doi:10.1002/humu.20159. PMID 15832313. S2CID 12500736.
- ^ Isaac, C.; Marsh, K. L.; Paznekas, W. A.; Dixon, J.; Dixon, M. J.; Jabs, E. W.; Meier, U. T. (September 2000). «Characterization of the nucleolar gene product, treacle, in Treacher Collins syndrome». Molecular Biology of the Cell. 11 (9): 3061–3071. doi:10.1091/mbc.11.9.3061. PMC 14975. PMID 10982400.
- ^ Gorlin RJ, Syndromes of the Head and Neck, 2001, Oxford University Press, 4th edition
- ^ a b Dixon, MJ; Marres, HA; Edwards, SJ; Dixon, J; Cremers, CW (April 1994). «Treacher Collins syndrome: correlation between clinical and genetic linkage studies». Clinical Dysmorphology. 3 (2): 96–103. doi:10.1097/00019605-199404000-00002. PMID 8055143.
- ^ Dixon, Jill; Ellis, Ian; Bottani, Armand; Temple, Karen; Dixon, Michael James (15 June 2004). «Identification of mutations in TCOF1: Use of molecular analysis in the pre- and postnatal diagnosis of Treacher Collins syndrome». American Journal of Medical Genetics. 127A (3): 244–248. doi:10.1002/ajmg.a.30010. PMID 15150774. S2CID 2091796.
- ^ Shoo, Brenda A.; McPherson, Elizabeth; Jabs, Ethylin Wang (1 April 2004). «Mosaicism of aTCOF1 mutation in an individual clinically unaffected with treacher collins syndrome». American Journal of Medical Genetics. 126A (1): 84–88. doi:10.1002/ajmg.a.20488. PMID 15039977. S2CID 35163245.
- ^ Splendore, Alessandra; Jabs, Ethylin Wang; Félix, Têmis Maria; Passos-Bueno, Maria Rita (31 August 2003). «Parental origin of mutations in sporadic cases of Treacher Collins syndrome». European Journal of Human Genetics. 11 (9): 718–722. doi:10.1038/sj.ejhg.5201029. PMID 12939661.
- ^ a b c d Senggen, E; Laswed, T; Meuwly, JY; Maestre, LA; Jaques, B; Meuli, R; Gudinchet, F (May 2011). «First and second branchial arch syndromes: multimodality approach» (PDF). Pediatric Radiology. 41 (5): 549–61. doi:10.1007/s00247-010-1831-3. PMID 20924574. S2CID 22416094. Archived (PDF) from the original on 2019-04-26. Retrieved 2020-01-15.
- ^ Vento AR, et al., The O.M.E.N.S classification of hemifacial microsomia, 1991, Cleft Palate Craniofac, J 28, p. 68-76
- ^ a b Posnick JC; et al. (2000). «Treacher Collins syndrome: current evaluation, treatment, and future directions». Cleft Palate Craniofac J. 55 (5): 1120–1133. doi:10.1597/1545-1569(2000)037<0434:TCSCET>2.0.CO;2. PMID 11034023.
- ^ Dixon, MJ (1995). «Treacher Collins syndrome». Journal of Medical Genetics. 32 (10): 806–8. doi:10.1136/jmg.32.10.806. PMC 1051706. PMID 8558560.
- ^ Goel L; et al. (2009). «Treacher Collins syndrome-a challenge for anaesthesiologists». Indian J Anaesth. 53 (4): 642–645. PMC 2894488. PMID 20640217.
- ^ Evans, Adele Karen; Rahbar, Reza; Rogers, Gary F.; Mulliken, John B.; Volk, Mark S. (31 May 2006). «Robin sequence: A retrospective review of 115 patients». International Journal of Pediatric Otorhinolaryngology. 70 (6): 973–980. doi:10.1016/j.ijporl.2005.10.016. PMID 16443284.
- ^ Rose, Edmund; Staats, Richard; Thissen, Ulrike; Otten, Jörg-Eland; Schmelzeisen, Rainer; Jonas, Irmtrud (1 August 2002). «Sleep-Related Obstructive Disordered Breathing in Cleft Palate Patients after Palatoplasty». Plastic and Reconstructive Surgery. 110 (2): 392–396. doi:10.1097/00006534-200208000-00002. PMID 12142649. S2CID 36499038.
- ^ Bannink, Natalja; Mathijssen, Irene M. J.; Joosten, Koen F. M. (1 September 2010). «Use of Ambulatory Polysomnography in Children With Syndromic Craniosynostosis». Journal of Craniofacial Surgery. 21 (5): 1365–1368. doi:10.1097/SCS.0b013e3181ec69a5. PMID 20856022. S2CID 43739792.
- ^ a b Marres, HA (2002). Hearing loss in the Treacher-Collins syndrome. Advances in Oto-rhino-laryngology. Vol. 61. pp. 209–15. doi:10.1159/000066811. ISBN 978-3-8055-7449-5. PMID 12408086.
- ^ a b Zhang, Zhiyong; Niu, Feng; Tang, Xiaojun; Yu, Bing; Liu, Jianfeng; Gui, Lai (1 September 2009). «Staged Reconstruction for Adult Complete Treacher Collins Syndrome». Journal of Craniofacial Surgery. 20 (5): 1433–1438. doi:10.1097/SCS.0b013e3181af21f9. PMID 19816274. S2CID 44847925.
- ^ Saadeh, Pierre B.; Chang, Christopher C.; Warren, Stephen M.; Reavey, Patrick; McCarthy, Joseph G.; Siebert, John W. (1 June 2008). «Microsurgical Correction of Facial Contour Deformities in Patients with Craniofacial Malformations: A 15-Year Experience». Plastic and Reconstructive Surgery. 121 (6): 368e–378e. doi:10.1097/PRS.0b013e3181707194. PMID 18520863. S2CID 27971712.
- ^ Argenta, Louis C.; Iacobucci, John J. (30 June 1989). «Treacher Collins Syndrome: Present concepts of the disorder and their surgical correction». World Journal of Surgery. 13 (4): 401–409. doi:10.1007/BF01660753. PMID 2773500. S2CID 27094477.
- ^ Marres, HA; Cremers, CW; Marres, EH (1995). «Treacher-Collins syndrome. Management of major and minor anomalies of the ear». Revue de Laryngologie – Otologie – Rhinologie. 116 (2): 105–108. PMID 7569369.
- ^ Jane Ridley (Aug 12, 2022). «Man’s parents rejected him at birth because of his appearance». Insider. Retrieved 2022-08-13.
- ^ Conte, Chiara; Maria Rosaria D’Apice; Fabrizio Rinaldi; Stefano Gambardella; Federica Sanguiuolo; Giuseppe Novelli (27 September 2011). «Novel mutations of TCOF1 gene in European patients with treacher Collins syndrome». Medical Genetics. 12: 125. doi:10.1186/1471-2350-12-125. PMC 3199234. PMID 21951868.
- ^ Treacher Collin E (1900). «Cases with symmetrical congenital notches in the outer part of each lid and defective development of the malar bones». Trans Ophthalmol Soc UK. 20: 190–192.
- ^ Franceschetti A, Klein D (1949). «Mandibulo-facial dysostosis: new hereditary syndrome». Acta Ophthalmol. 27: 143–224.
- ^ Jr, Donald G. McNeil (1977-07-26). «Surgical Teamwork Gives Disease Victims a New Life». The New York Times. ISSN 0362-4331. Retrieved 2023-01-25.
- ^ «Bob Saget Biography». Retrieved 2022-06-30.
{{cite web}}: CS1 maint: url-status (link) - ^ «Nip/Tuck: Blu Mondae — TV.com». Archived from the original on 2008-06-12. Retrieved 2008-05-12.
- ^ a b «First Coast News: Local Family Has Daughter Born Without a Face».
- ^ «BBC programme page for Love Me, Love My Face». BBC Three. 17 June 2011. Archived from the original on 21 January 2018. Retrieved 7 November 2017.
- ^ «BBC programme page for So What If My Baby…» BBC Three. 24 August 2011. Archived from the original on 2 June 2017. Retrieved 7 November 2017.
- ^ «BBC Three Bringing Up Britain season». BBC One. 12 April 2011. Archived from the original on 12 August 2011. Retrieved 24 August 2011.
- ^ «Finding My Family on Facebook». bbc.co.uk. BBC Three. 2011. Archived from the original on 2011-10-31..
- ^ Chilton, Martin (24 February 2012). «Wonder by R. J. Palacio: review». The Telegraph. Archived from the original on 6 October 2017. Retrieved 7 November 2017.
- ^ «Julia Roberts’ Drama ‘Wonder’ Pushed to November». The Hollywood Reporter. February 13, 2017. Archived from the original on April 22, 2017. Retrieved February 13, 2017.
- ^ O’Conner, Katie (December 22, 2017). «Real Life Reflected on the Silver Screen». Richmond Times-Dispatch.
- ^ Wilner, Norman (February 18, 2020). «Canadian Screen Awards 2020: Prepare for a Schitt’s show». Now. Archived from the original on 2020-02-18. Retrieved December 28, 2020.
External links[edit]
| Treacher Collins syndrome | |
|---|---|
| Other names | Treacher Collins–Franceschetti syndrome,[1] mandibulofacial dysostosis,[2] Franceschetti-Zwalen-Klein syndrome[3] |
|
|
| Child with Treacher Collins syndrome[4] | |
| Specialty | Medical genetics |
| Symptoms | Deformities of the ears, eyes, cheekbones, chin[5] |
| Complications | Breathing problems, problems seeing, hearing loss[5] |
| Causes | Genetic[5] |
| Diagnostic method | Based on symptoms, X-rays, genetic testing[3] |
| Differential diagnosis | Nager syndrome, Miller syndrome, hemifacial microsomia[3] |
| Treatment | Reconstructive surgery, hearing aids, speech therapy[6] |
| Prognosis | Generally normal life expectancy[6] |
| Frequency | 1 in 50,000 people[5] |
Treacher Collins syndrome (TCS) is a genetic disorder characterized by deformities of the ears, eyes, cheekbones, and chin.[5] The degree to which a person is affected, however, may vary from mild to severe.[5] Complications may include breathing problems, problems seeing, cleft palate, and hearing loss.[5] Those affected generally have normal intelligence.[5]
TCS is usually autosomal dominant.[5] More than half the time it occurs as a result of a new mutation rather than being inherited.[5] The involved genes may include TCOF1, POLR1C, or POLR1D.[5] Diagnosis is generally suspected based on symptoms and X-rays, and potentially confirmation by genetic testing.[3]
Treacher Collins syndrome is not curable.[6] Symptoms may be managed with reconstructive surgery, hearing aids, speech therapy, and other assistive devices.[6] Life expectancy is generally normal.[6] TCS occurs in about one in 50,000 people.[5] The syndrome is named after Edward Treacher Collins, an English surgeon and ophthalmologist, who described its essential traits in 1900.[7][8]
Signs and symptoms[edit]
The same child shown from the front above in infobox, now seen from the side, with small ears and a chin that is far back.[4]
Symptoms in people with Treacher Collins syndrome vary. Some individuals are so mildly affected that they remain undiagnosed, while others have moderate to severe facial involvement and life-threatening airway compromise.[9] Most of the features of TCS are symmetrical and are already recognizable at birth.[citation needed]
The most common symptom of Treacher Collins syndrome is underdevelopment of the lower jaw and underdevelopment of the zygomatic bone. This can be accompanied by the tongue being retracted. The small mandible can result in a poor occlusion of the teeth or in more severe cases, trouble breathing or swallowing. The respiratory system of a child with Treacher Collins syndrome is the primary concern at birth, with other issues only addressed once respiratory function has been stabilized.[10] Underdevelopment of the zygomatic bone gives the cheeks a sunken appearance.[11][12]
The external ear is sometimes small, rotated, malformed, or absent entirely in people with TCS. Symmetric, bilateral narrowing or absence of the external ear canal, is also described.[12][13] In most cases, the bones of the middle ear and the middle ear cavity are misshapen. Inner ear malformations are rarely described. As a result of these abnormalities, a majority of the individuals with TCS have conductive hearing loss.[12][14]
Most affected people also experience eye problems, including coloboma (notches) in the lower eyelids, partial or complete absence of eyelashes on the lower lid, downward angled eyelids, drooping of upper and lower eyelids, and narrowing of the tear ducts. Vision loss can occur and is associated with strabismus, refractive errors, and anisometropia. It can also be caused by severely dry eyes, a consequence of lower eyelid abnormalities and frequent eye infections.[12][13][15][16]
Although an abnormally shaped skull is not distinctive for Treacher Collins syndrome, brachycephaly with bitemporal narrowing is sometimes observed.[13] Cleft palate is also common.[12]
Dental anomalies are seen in 60% of affected people, including tooth agenesis (33%), discoloration (enamel opacities) (20%), malplacement of the maxillary first molars (13%), and wide spacing of the teeth. In some cases, dental anomalies in combination with mandible hypoplasia result in a malocclusion. This can lead to problems with food intake and the ability to close the mouth.[12]
Less common features of TCS may add to an affected person’s breathing problems, including sleep apnea. Choanal atresia or stenosis is a narrowing or absence of the choanae, the internal opening of the nasal passages, which may also be observed. Underdevelopment of the pharynx can also narrow the airway.[12]
Features related to TCS that are seen less frequently include nasal deformities, high-arched palate, macrostomia, preauricular hair displacement, cleft palate, hypertelorism, notched upper eyelid, and congenital heart defects.[11][12][16]
Although facial deformity is often associated with developmental delay and intellectual disability, more than 95% of people affected with TCS have normal intelligence.[12] The psychological and social problems associated with facial deformity can affect quality of life in individuals with TCS.
Genetics[edit]
Mutations in TCOF1, POLR1C, or POLR1D genes can cause Treacher Collins syndrome.[17] TCOF1 gene mutations are the most common cause of the disorder, with POLR1C and POLR1D gene mutations causing an additional 2% of cases. In individuals without an identified mutation in one of these genes, the genetic cause of the condition is unknown. The TCOF1, POLR1C, and POLR1D genes code for proteins which play important roles in the early development of bones and other tissues of the face. Mutations in these genes reduce the production of rRNA, which may trigger the self-destruction (apoptosis) of certain cells involved in the development of facial bones and tissues. It is unclear why the effects of a reduction in rRNA are limited to facial development. Mutations in TCOF1 and POLR1D cause the autosomal dominant form of Treacher Collins, and mutations in POLR1C cause the autosomal recessive form.[12]
TCOF1[edit]
TCOF1 is the primary gene associated with TCS, a mutation in this gene being found in 90–95% of the individuals with TCS.[11][18] However, in some individuals with typical symptoms of TCS, mutations in TCOF1 have not been found.[19] Investigation of the DNA has resulted in the identification of the kind of mutations found in TCOF1. The majority of mutations are small deletions or insertions, though splice site and missense mutations also have been identified.[11][20][21][22]
Mutation analysis has unveiled more than 100 disease-causing mutations in TCOF1, which are mostly family-specific mutations. The only recurrent mutation accounts for about 17% of the cases.[23]
TCOF1 is found on the 5th chromosome in the 5q32 region. It codes for a relatively simple nucleolar protein called treacle, that is thought to be involved in ribosome assembly.[18] Mutations in TCOF1 lead to haploinsufficiency of the treacle protein.[24] Haploinsufficiency occurs when a diploid organism has only one functional copy of a gene, because the other copy is inactivated by a mutation. The one normal copy of the gene does not produce enough protein, causing disease. Haploinsufficiency of the treacle protein leads to a depletion of the neural crest cell precursor, which leads to a reduced number of crest cells migrating to the first and second pharyngeal arches. These cells play an important role in the development of the craniofacial appearance, and loss of one copy of treacle affects the cells’ ability to form the bones and tissues of the face.[12][20][25]
Other mutations[edit]
POLR1C and POLR1D mutations are responsible for a minority of cases of Treacher Collins. POLR1C is found on chromosome 6 at position 6q21.2 and POLR1D is found on chromosome 13 at position 13q12.2. Those genes code for a protein subunits shared between RNA polymerase I and III. Both of these polymerases are important for ribosome biogenesis.[12]
Diagnosis[edit]
Genetic counseling[edit]
TCS is inherited in an autosomal dominant manner and the penetrance of the affected gene is almost complete.[26] Some recent investigations, though, described some rare cases in which the penetrance in TCS was not complete. Causes may be a variable expressivity, an incomplete penetrance[27] or germline mosaicism.[28] Only 40% of the mutations are inherited. The remaining 60% are a result of a de novo mutation, where a child has a new mutation in the responsible gene and did not inherit it from either parent.[12][29] In the outcome of the disease, inter- and intrafamilial variability occurs. This suggests that when an affected child is born, it is important to investigate the parents to determine whether the affected gene is present, because the parent could have a mild form of the disease that has not been diagnosed. In this case, the risk of having another affected child is 50%. If the parents do not have the affected gene, the recurrence risk appears to be low.[26] In following generations, the severity of the clinical symptoms increases.[21]
Prenatal diagnosis[edit]
Mutations in the main genes responsible for TCS can be detected with chorionic villus sampling or amniocentesis. Rare mutations may not be detected by these methods. Ultrasonography can be used to detect craniofacial abnormalities later in pregnancy, but may not detect milder cases.[12]
Clinical findings[edit]
TCS is often first suspected with characteristic symptoms observed during a physical exam. However, the clinical presentation of TCS can resemble other diseases, making diagnosis difficult.[30] The OMENS classification was developed as a comprehensive and stage-based approach to differentiate the diseases. This acronym describes five distinct dysmorphic manifestations, namely orbital asymmetry, mandibular hypoplasia, auricular deformity, nerve development, and soft-tissue disease.[31]
Orbital symmetry
- O0: normal orbital size, position
- O1: abnormal orbital size
- O2: abnormal orbital position
- O3: abnormal orbital size and position
Mandible
- M0: normal mandible
- M1: small mandible and glenoid fossa with short ramus
- M2: ramus short and abnormally shaped
- 2A: glenoid fossa in anatomical acceptable position
- 2B: Temperomandibular joint inferiorly (TMJ), medially, anteriorly displaced, with severely hypoplastic condyle
- M3: Complete absence of ramus, glenoid fossa, and TMJ
Ear
- E0: normal ear
- E1: Minor hypoplasia and cupping with all structures present
- E2: Absence of external auditory canal with variable hypoplasia of the auricle
- E3: Malposition of the lobule with absent auricle, lobular remnant usually inferior anteriorly displaced
Facial nerve
- N0: No facial nerve involvement
- N1: Upper facial nerve involvement (temporal or zygomatic branches)
- N2: Lower facial nerve involvement (buccal, mandibular or cervical)
- N3: All branches affected
Soft tissue
- S0: No soft tissue or muscle deficiency
- S1: Minimal tissue or muscle deficiency
- S2: Moderate tissue or muscle deficiency
- S3: Severe tissue or muscle deficiency
Radiographs[edit]
A few techniques are used to confirm the diagnosis in TCS.[30][32]
An orthopantomogram (OPG) is a panoramic dental X-ray of the upper and lower jaw. It shows a two-dimensional image from ear to ear. Particularly, OPG facilitates an accurate postoperative follow-up and monitoring of bone growth under a mono- or double-distractor treatment. Thereby, some TCS features could be seen on OPG, but better techniques are used to include the whole spectrum of TCS abnormalities instead of showing only the jaw abnormalities.[30]
Another method of radiographic evaluation is taking an X-ray image of the whole head. The lateral cephalometric radiograph in TCS shows hypoplasia of the facial bones, like the malar bone, mandible, and the mastoid.[30]
Finally, occipitomental radiographs are used to detect hypoplasia or discontinuity of the zygomatic arch.[32]
CT scan[edit]
A temporal-bone CT using thin slices makes it possible to diagnose the degree of stenosis and atresia of the external auditory canal, the status of the middle ear cavity, the absent or dysplastic and rudimentary ossicles, or inner ear abnormalities such as a deficient cochlea. Two- and three-dimensional CT reconstructions with VRT and bone and skin-surfacing are helpful for more accurate staging and the three-dimensional planning of mandibular and external ear reconstructive surgery.[citation needed]
Differential diagnosis[edit]
Other diseases have similar characteristics to Treacher Collins syndrome. In the differential diagnosis, one should consider the acrofacial dysostoses. The facial appearance resembles that of Treacher Collins syndrome, but additional limb abnormalities occur in those persons. Examples of these diseases are Nager syndrome and Miller syndrome.[citation needed]
The oculoauriculovertebral spectrum should also be considered in the differential diagnosis. An example is hemifacial microsomia, which primarily affects development of the ear, mouth, and mandible. This anomaly may occur bilaterally. Another disease which belongs to this spectrum is Goldenhar syndrome, which includes vertebral abnormalities, epibulbar dermoids and facial deformities.[33]
Treatment[edit]
The treatment of individuals with TCS may involve the intervention of professionals from multiple disciplines. The primary concerns are breathing and feeding, as a consequence of the hypoplasia of the mandibula and the obstruction of the hypopharynx by the tongue. Sometimes, they may require a tracheostomy to maintain an adequate airway,[34] and a gastrostomy to assure an adequate caloric intake while protecting the airway. Corrective surgery of the face is performed at defined ages, depending on the developmental state.[35]
An overview of the present guidelines:
- If a cleft palate is present, the repair normally takes place at 9–12 months old. Before surgery, a polysomnography with a palatal plate in place is needed. This may predict the postoperative situation and gives insight on the chance of the presence of sleep apnea (OSAS) after the operation.[11][36][37]
- Hearing loss is treated by bone conduction amplification, speech therapy, and educational intervention to avoid language/speech problems. The bone-anchored hearing aid is an alternative for individuals with ear anomalies.[38]
- Zygomatic and orbital reconstruction is performed when the cranio-orbitozygomatic bone is completely developed, usually at the age of 5–7 years. In children, an autologous bone graft is mostly used. In combination with this transplantation, lipofilling can be used in the periorbital area to get an optimal result of the reconstruction.[citation needed] Reconstruction of the lower eyelid coloboma includes the use of a myocutaneous flap, which is elevated and in this manner closes the eyelid defect.[39]
- External ear reconstruction is usually done when the individual is at least eight years old. Sometimes, the external auditory canal or middle ear can also be treated.
- The optimal age for the maxillomandibular reconstruction is controversial; as of 2004, this classification has been used:[11]
- Type I (mild) and Type IIa (moderate) 13–16 years
- Type IIb (moderate to severe malformation) at skeletal maturity
- Type III (severe) 6–10 years
- When the teeth are cutting, the teeth should be under supervision of an orthodontist to make sure no abnormalities occur. If abnormalities like dislocation or an overgrowth of teeth are seen, appropriate action can be undertaken as soon as possible.[20]
- Orthognathic treatments usually take place after the age of 16 years; at this point, all teeth are in place and the jaw and dentition are mature. Whenever OSAS is detected, the level of obstruction is determined through endoscopy of the upper airways. Mandibular advancement can be an effective way to improve both breathing and æsthetics, while a genioplasty only restores the profile.[11]
- If a nose reconstruction is necessary, it is usually performed after the orthognathic surgery and after the age of 18 years.[11]
- The contour of the facial soft tissues generally requires correction at a later age, because of the facial skeletal maturity. The use of microsurgical methods, like the free flap transfer, has improved the correction of facial soft tissue contours.[40] Another technique to improve the facial soft tissue contours is lipofilling. For instance, lipofilling is used to reconstruct the eyelids.[39]
Hearing loss[edit]
Hearing loss in Treacher Collins syndrome is caused by deformed structures in the outer and middle ear. The hearing loss is generally bilateral with a conductive loss of about 50–70 dB. Even in cases with normal auricles and open external auditory canals, the ossicular chain is often malformed.[41]
Attempts to surgically reconstruct the external auditory canal and improve hearing in children with TCS have not yielded positive results.[42]
Auditory rehabilitation with bone-anchored hearing aids (BAHAs) or a conventional bone conduction aid has proven preferable to surgical reconstruction.[38]
Psychiatric[edit]
The disorder can be associated with a number of psychological symptoms, including anxiety, depression, social phobia, and distress about body image. People who have this disorder may also experience discrimination, bullying, and name calling, especially when young. A multi-disciplinary team and parental support should include these issues.[43]
Epidemiology[edit]
TCS occurs in about one in 50,000 births in Europe.[44] Worldwide, it is estimated to occur in one in 10,000 to one in 50,000 births.[12]
History[edit]
The syndrome is named after Edward Treacher Collins (1862–1932), the English surgeon and ophthalmologist who described its essential traits in 1900.[7][8][45] In 1949, Adolphe Franceschetti and David Klein described the same condition on their own observations as mandibulofacial dysostosis. The term mandibulofacial dysostosis is used to describe the clinical features.[46]
Culture[edit]
In July 1977, a New York Times article describing new plastic surgery techniques which could partially correct the appearance of those with Treacher Collins syndrome was widely circulated resulting in raised awareness of the disease.[47]
Prior to beginning his comedy career, Bob Saget made a documentary short called «Through Adam’s Eyes» documenting his young nephew’s experiences undergoing facial reconstructive surgery due to Treacher Collins; the film won a student Academy Award.[48]
The disorder was featured on the show Nip/Tuck, in the episode «Blu Mondae».[49] TLC’s Born Without a Face[50] features Juliana Wetmore, who was born with the most severe case in medical history of this syndrome and is missing 30%–40% of the bones in her face.[50]
In 2010, BBC Three documentary Love Me, Love My Face[51] covered the case of a man, Jono Lancaster, with the condition. In 2011, BBC Three returned to Jono to cover his and his partner Laura’s quest to start a family,[2] in So What If My Baby Is Born Like Me?,[52] which first aired as part of a BBC Three season of programmes on parenting.[53] The first film was replayed on BBC One shortly ahead of the second film’s initial BBC Three broadcast. Lancaster’s third BBC Three film, Finding My Family on Facebook, which looked at adoption, aired in 2011.[54]
In Wonder, a children’s novel, the main character is a child who has Treacher Collins syndrome.[55] A 2017 film adaptation of Wonder, starring Julia Roberts, Owen Wilson and Jacob Tremblay, was released in November 2017.[56][57]
Alison Midstokke, who appears in the drama film Happy Face (2018),[58] is an actress and activist who has the condition.
See also[edit]
- First arch syndrome
- Franceschetti-Klein syndrome
- Hearing loss with craniofacial syndromes
References[edit]
- ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. pp. 894, 1686. ISBN 978-1-4160-2999-1.
- ^ a b «I hated seeing my face in the mirror». BBC Online. 18 November 2010. Archived from the original on 2018-10-11. Retrieved 2010-11-18.
- ^ a b c d «Treacher Collins Syndrome». NORD (National Organization for Rare Disorders). 2016. Archived from the original on 20 December 2019. Retrieved 7 November 2017.
- ^ a b Goel, L; Bennur, SK; Jambhale, S (August 2009). «Treacher-collins syndrome-a challenge for anaesthesiologists». Indian Journal of Anaesthesia. 53 (4): 496–500. PMC 2894488. PMID 20640217.
- ^ a b c d e f g h i j k l «Treacher Collins syndrome». Genetics Home Reference. June 2012. Archived from the original on 26 June 2020. Retrieved 7 November 2017.
- ^ a b c d e «Treacher Collins syndrome». rarediseases.info.nih.gov. 2015. Archived from the original on 16 August 2019. Retrieved 7 November 2017.
- ^ a b R, Pramod John; John, Pramod (2014). Textbook of Oral Medicine. JP Medical Ltd. p. 76. ISBN 9789350908501. Archived from the original on 2020-09-12. Retrieved 2017-09-17.
- ^ a b Beighton, Greta (2012). The Man Behind the Syndrome. Springer Science & Business Media. p. 173. ISBN 9781447114154.
- ^ Edwards, S J; Fowlie, A; Cust, M P; Liu, D T; Young, I D; Dixon, M J (1 July 1996). «Prenatal diagnosis in Treacher Collins syndrome using combined linkage analysis and ultrasound imaging». Journal of Medical Genetics. 33 (7): 603–606. doi:10.1136/jmg.33.7.603. PMC 1050672. PMID 8818950.
- ^ Mouthon, L., Busa, T., Bretelle, F., Karmous‐Benailly, H., Missirian, C., Philip, N., & Sigaudy, S. (2019). Prenatal diagnosis of micrognathia in 41 fetuses: Retrospective analysis of outcome and genetic etiologies. American Journal of Medical Genetics Part A, 179(12), 2365–2373. doi: 10.1002/ajmg.a.61359
- ^ a b c d e f g h Katsanis SH, et al., Treacher Collins syndrome, 2004, GeneReviews
- ^ a b c d e f g h i j k l m n o «The Physician’s Guide to Treacher Collins Syndrome» (PDF). National Organization for Rare Disorders (NORD). 2012. Archived from the original (PDF) on 2017-01-28.
- ^ a b c Posnick, Jeffrey C (1 October 1997). «Treacher Collins syndrome: Perspectives in evaluation and treatment». Journal of Oral and Maxillofacial Surgery. 55 (10): 1120–1133. doi:10.1016/S0278-2391(97)90294-9. PMID 9331237.
- ^ Trainor, Paul A; Dixon, Jill; Dixon, Michael J (24 December 2008). «Treacher Collins syndrome: etiology, pathogenesis and prevention». European Journal of Human Genetics. 17 (3): 275–283. doi:10.1038/ejhg.2008.221. PMC 2986179. PMID 19107148.
- ^ Hertle, R W; Ziylan, S; Katowitz, J A (1 October 1993). «Ophthalmic features and visual prognosis in the Treacher-Collins syndrome». British Journal of Ophthalmology. 77 (10): 642–645. doi:10.1136/bjo.77.10.642. PMC 504607. PMID 8218033.
- ^ a b Marszałek, B; Wójcicki, P; Kobus, K; Trzeciak, WH (2002). «Clinical features, treatment and genetic background of Treacher Collins syndrome». Journal of Applied Genetics. 43 (2): 223–33. PMID 12080178.
- ^ «Treacher Collins Syndrome». NORD (National Organization for Rare Disorders). Archived from the original on 2019-12-20. Retrieved 2016-02-29.
- ^ a b Dixon, Jill; Edwards, Sara J.; Gladwin, Amanda J.; Dixon, Michael J.; Loftus, Stacie K.; Bonner, Cynthia A.; Koprivnikar, Kathryn; Wasmuth, John J. (31 January 1996). «Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome». Nature Genetics. 12 (2): 130–136. doi:10.1038/ng0296-130. PMID 8563749. S2CID 34312227.
- ^ Teber OA, Gillessen-Kaesbach G, Fischer S, Böhringer S, Albrecht B, Albert A, Arslan-Kirchner M, Haan E, Hagedorn-Greiwe M, Hammans C, Henn W, Hinkel GK, König R, Kunstmann E, Kunze J, Neumann LM, Prott EC, Rauch A, Rott HD, Seidel H, Spranger S, Sprengel M, Zoll B, Lohmann DR, Wieczorek D (2004). «Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation». European Journal of Human Genetics. 12 (11): 879–90. doi:10.1038/sj.ejhg.5201260. PMID 15340364.
- ^ a b c Dixon, J; Trainor, P.; Dixon, M. J. (1 May 2007). «Treacher Collins syndrome». Orthodontics & Craniofacial Research. 10 (2): 88–95. doi:10.1111/j.1601-6343.2007.00388.x. PMID 17552945.
- ^ a b Masotti, Cibele; Ornelas, Camila C.; Splendore-Gordonos, Alessandra; Moura, Ricardo; Félix, Têmis M.; Alonso, Nivaldo; Camargo, Anamaria A.; Passos-Bueno, Maria (1 January 2009). «Reduced transcription of TCOF1 in adult cells of Treacher Collins syndrome patients». BMC Medical Genetics. 10 (1): 136. doi:10.1186/1471-2350-10-136. PMC 2801500. PMID 20003452.
- ^ Sakai, Daisuke; Trainor, Paul A. (31 May 2009). «Treacher Collins syndrome: Unmasking the role of Tcof1/treacle». The International Journal of Biochemistry & Cell Biology. 41 (6): 1229–1232. doi:10.1016/j.biocel.2008.10.026. PMC 3093759. PMID 19027870.
- ^ Splendore, Alessandra; Fanganiello, Roberto D.; Masotti, Cibele; Morganti, Lucas S. C.; Rita Passos-Bueno, M. (1 May 2005). «TCOF1 mutation database: Novel mutation in the alternatively spliced exon 6A and update in mutation nomenclature». Human Mutation. 25 (5): 429–434. doi:10.1002/humu.20159. PMID 15832313. S2CID 12500736.
- ^ Isaac, C.; Marsh, K. L.; Paznekas, W. A.; Dixon, J.; Dixon, M. J.; Jabs, E. W.; Meier, U. T. (September 2000). «Characterization of the nucleolar gene product, treacle, in Treacher Collins syndrome». Molecular Biology of the Cell. 11 (9): 3061–3071. doi:10.1091/mbc.11.9.3061. PMC 14975. PMID 10982400.
- ^ Gorlin RJ, Syndromes of the Head and Neck, 2001, Oxford University Press, 4th edition
- ^ a b Dixon, MJ; Marres, HA; Edwards, SJ; Dixon, J; Cremers, CW (April 1994). «Treacher Collins syndrome: correlation between clinical and genetic linkage studies». Clinical Dysmorphology. 3 (2): 96–103. doi:10.1097/00019605-199404000-00002. PMID 8055143.
- ^ Dixon, Jill; Ellis, Ian; Bottani, Armand; Temple, Karen; Dixon, Michael James (15 June 2004). «Identification of mutations in TCOF1: Use of molecular analysis in the pre- and postnatal diagnosis of Treacher Collins syndrome». American Journal of Medical Genetics. 127A (3): 244–248. doi:10.1002/ajmg.a.30010. PMID 15150774. S2CID 2091796.
- ^ Shoo, Brenda A.; McPherson, Elizabeth; Jabs, Ethylin Wang (1 April 2004). «Mosaicism of aTCOF1 mutation in an individual clinically unaffected with treacher collins syndrome». American Journal of Medical Genetics. 126A (1): 84–88. doi:10.1002/ajmg.a.20488. PMID 15039977. S2CID 35163245.
- ^ Splendore, Alessandra; Jabs, Ethylin Wang; Félix, Têmis Maria; Passos-Bueno, Maria Rita (31 August 2003). «Parental origin of mutations in sporadic cases of Treacher Collins syndrome». European Journal of Human Genetics. 11 (9): 718–722. doi:10.1038/sj.ejhg.5201029. PMID 12939661.
- ^ a b c d Senggen, E; Laswed, T; Meuwly, JY; Maestre, LA; Jaques, B; Meuli, R; Gudinchet, F (May 2011). «First and second branchial arch syndromes: multimodality approach» (PDF). Pediatric Radiology. 41 (5): 549–61. doi:10.1007/s00247-010-1831-3. PMID 20924574. S2CID 22416094. Archived (PDF) from the original on 2019-04-26. Retrieved 2020-01-15.
- ^ Vento AR, et al., The O.M.E.N.S classification of hemifacial microsomia, 1991, Cleft Palate Craniofac, J 28, p. 68-76
- ^ a b Posnick JC; et al. (2000). «Treacher Collins syndrome: current evaluation, treatment, and future directions». Cleft Palate Craniofac J. 55 (5): 1120–1133. doi:10.1597/1545-1569(2000)037<0434:TCSCET>2.0.CO;2. PMID 11034023.
- ^ Dixon, MJ (1995). «Treacher Collins syndrome». Journal of Medical Genetics. 32 (10): 806–8. doi:10.1136/jmg.32.10.806. PMC 1051706. PMID 8558560.
- ^ Goel L; et al. (2009). «Treacher Collins syndrome-a challenge for anaesthesiologists». Indian J Anaesth. 53 (4): 642–645. PMC 2894488. PMID 20640217.
- ^ Evans, Adele Karen; Rahbar, Reza; Rogers, Gary F.; Mulliken, John B.; Volk, Mark S. (31 May 2006). «Robin sequence: A retrospective review of 115 patients». International Journal of Pediatric Otorhinolaryngology. 70 (6): 973–980. doi:10.1016/j.ijporl.2005.10.016. PMID 16443284.
- ^ Rose, Edmund; Staats, Richard; Thissen, Ulrike; Otten, Jörg-Eland; Schmelzeisen, Rainer; Jonas, Irmtrud (1 August 2002). «Sleep-Related Obstructive Disordered Breathing in Cleft Palate Patients after Palatoplasty». Plastic and Reconstructive Surgery. 110 (2): 392–396. doi:10.1097/00006534-200208000-00002. PMID 12142649. S2CID 36499038.
- ^ Bannink, Natalja; Mathijssen, Irene M. J.; Joosten, Koen F. M. (1 September 2010). «Use of Ambulatory Polysomnography in Children With Syndromic Craniosynostosis». Journal of Craniofacial Surgery. 21 (5): 1365–1368. doi:10.1097/SCS.0b013e3181ec69a5. PMID 20856022. S2CID 43739792.
- ^ a b Marres, HA (2002). Hearing loss in the Treacher-Collins syndrome. Advances in Oto-rhino-laryngology. Vol. 61. pp. 209–15. doi:10.1159/000066811. ISBN 978-3-8055-7449-5. PMID 12408086.
- ^ a b Zhang, Zhiyong; Niu, Feng; Tang, Xiaojun; Yu, Bing; Liu, Jianfeng; Gui, Lai (1 September 2009). «Staged Reconstruction for Adult Complete Treacher Collins Syndrome». Journal of Craniofacial Surgery. 20 (5): 1433–1438. doi:10.1097/SCS.0b013e3181af21f9. PMID 19816274. S2CID 44847925.
- ^ Saadeh, Pierre B.; Chang, Christopher C.; Warren, Stephen M.; Reavey, Patrick; McCarthy, Joseph G.; Siebert, John W. (1 June 2008). «Microsurgical Correction of Facial Contour Deformities in Patients with Craniofacial Malformations: A 15-Year Experience». Plastic and Reconstructive Surgery. 121 (6): 368e–378e. doi:10.1097/PRS.0b013e3181707194. PMID 18520863. S2CID 27971712.
- ^ Argenta, Louis C.; Iacobucci, John J. (30 June 1989). «Treacher Collins Syndrome: Present concepts of the disorder and their surgical correction». World Journal of Surgery. 13 (4): 401–409. doi:10.1007/BF01660753. PMID 2773500. S2CID 27094477.
- ^ Marres, HA; Cremers, CW; Marres, EH (1995). «Treacher-Collins syndrome. Management of major and minor anomalies of the ear». Revue de Laryngologie – Otologie – Rhinologie. 116 (2): 105–108. PMID 7569369.
- ^ Jane Ridley (Aug 12, 2022). «Man’s parents rejected him at birth because of his appearance». Insider. Retrieved 2022-08-13.
- ^ Conte, Chiara; Maria Rosaria D’Apice; Fabrizio Rinaldi; Stefano Gambardella; Federica Sanguiuolo; Giuseppe Novelli (27 September 2011). «Novel mutations of TCOF1 gene in European patients with treacher Collins syndrome». Medical Genetics. 12: 125. doi:10.1186/1471-2350-12-125. PMC 3199234. PMID 21951868.
- ^ Treacher Collin E (1900). «Cases with symmetrical congenital notches in the outer part of each lid and defective development of the malar bones». Trans Ophthalmol Soc UK. 20: 190–192.
- ^ Franceschetti A, Klein D (1949). «Mandibulo-facial dysostosis: new hereditary syndrome». Acta Ophthalmol. 27: 143–224.
- ^ Jr, Donald G. McNeil (1977-07-26). «Surgical Teamwork Gives Disease Victims a New Life». The New York Times. ISSN 0362-4331. Retrieved 2023-01-25.
- ^ «Bob Saget Biography». Retrieved 2022-06-30.
{{cite web}}: CS1 maint: url-status (link) - ^ «Nip/Tuck: Blu Mondae — TV.com». Archived from the original on 2008-06-12. Retrieved 2008-05-12.
- ^ a b «First Coast News: Local Family Has Daughter Born Without a Face».
- ^ «BBC programme page for Love Me, Love My Face». BBC Three. 17 June 2011. Archived from the original on 21 January 2018. Retrieved 7 November 2017.
- ^ «BBC programme page for So What If My Baby…» BBC Three. 24 August 2011. Archived from the original on 2 June 2017. Retrieved 7 November 2017.
- ^ «BBC Three Bringing Up Britain season». BBC One. 12 April 2011. Archived from the original on 12 August 2011. Retrieved 24 August 2011.
- ^ «Finding My Family on Facebook». bbc.co.uk. BBC Three. 2011. Archived from the original on 2011-10-31..
- ^ Chilton, Martin (24 February 2012). «Wonder by R. J. Palacio: review». The Telegraph. Archived from the original on 6 October 2017. Retrieved 7 November 2017.
- ^ «Julia Roberts’ Drama ‘Wonder’ Pushed to November». The Hollywood Reporter. February 13, 2017. Archived from the original on April 22, 2017. Retrieved February 13, 2017.
- ^ O’Conner, Katie (December 22, 2017). «Real Life Reflected on the Silver Screen». Richmond Times-Dispatch.
- ^ Wilner, Norman (February 18, 2020). «Canadian Screen Awards 2020: Prepare for a Schitt’s show». Now. Archived from the original on 2020-02-18. Retrieved December 28, 2020.
External links[edit]
Генетические нарушения у человека и методы их выявления

Генами называются участки ДНК, в которых закодирована структура всех белков в теле человека или любого другого живого организма. В биологии действует правило: «один ген – один белок», то есть в каждом гене содержится информация только об одном определенном белке.
В 1990 году большая группа ученых из разных стран начала проект под названием «Геном человека». Он завершился в 2003 году и помог установить, что человеческий геном содержит 20–25 тысяч генов. Каждый ген представлен двумя копиями, которые кодируют один и тот же белок, но могут немного различаться. Большинство генов одинаковые у всех людей – различается всего 1%.
ДНК находится в клетке внутри ядра. Она особым образом организована в виде хромосом – эти нитеподобные структуры можно рассмотреть в микроскоп с достаточно большим увеличением. Внутри хромосомы ДНК намотана на белки – гистоны. Когда гены неактивны, они расположены очень компактно, а во время считывания генетического материала молекула ДНК расплетается.
В клетках человека есть структуры, которые называются митохондриями. Они выполняют роль «электростанций» и отвечают за дыхание. Это единственные клеточные органеллы, у которых есть собственная ДНК. И в ней тоже могут возникать нарушения.

Весь набор хромосом в клетке называется кариотипом. В норме у человека он представлен 23 парами хромосом, всего их 46. Выделяют два вида хромосом:
- 22 пары аутосом одинаковы у мужчин и женщин. В каждой паре хромосомы имеют одинаковую длину и содержат одинаковые наборы генов.
- Одна пара половых хромосом. У женщин это две X-хромосомы. Одна из них неактивна и плотно свернута – ее называют тельцем Барра. У мужчин одна половая хромосома представлена X-хромосомой, а вторая – Y-хромосомой, она меньше по размерам.
Методы исследования хромосом
Для исследования кариотипа применяют специальный метод – световую микроскопию дифференциально окрашенных метафазных хромосом культивированных лимфоцитов периферической крови.
Этот анализ применяется для диагностики различных хромосомных заболеваний. Он позволяет выявлять такие нарушения, как:
- Грубые изменения в кариотипе – изменение количества хромосом. Например, при синдроме Дауна в клетках ребенка присутствует лишняя хромосома №21.
- Присутствие в организме клеток с разными кариотипами. Это явление называется мозаицизмом.
- Хромосомные аберрации – нарушение структуры хромосом, внутрихромосомные и межхромосомные перестройки. Сюда относятся делеции (утрата участка хромосомы), дупликации (удвоение участка хромосомы), инверсии (поворот участка хромосомы на 180 градусов), транслокации (перенос участка одной хромосомы в другую).

Однако с помощью исследования кариотипа можно выявить не все генетические нарушения. Оно не способно обнаружить такие изменения, как:
- микроделеции и микродупликации, когда утрачивается или дублируется очень маленький участок хромосомы;
- болезни обмена, вызванные нарушением последовательности «букв» генетического кода в отдельных генах;
- митохондриальные заболевания, связанные с нарушениями в генетическом материале митохондрий;
- низкопроцентный мозаицизм, когда клеток с неправильным кариотипом очень мало;
- мутации в отдельных генах, которые не приводят к изменению внешнего вида хромосом;
- эпигенетические расстройства, при которых структура хромосом и генов не меняется, но изменяется их функция.

Для получения дополнительной информации, не видимой в световой микроскоп, используют хромосомный микроматричный анализ (ХМА). С его помощью можно изучить все клинически значимые участки генома и выявить изменения в количестве и структуре хромосом, а именно микрополомки (микроделеции и микродупликации).
Во время хромосомного микроматричного анализа применяют технологию полногеномной амплификации и гибридизации фрагментов опытной ДНК с олигонуклеотидами, нанесенными на микроматрицу. Если объяснять простыми словами, то сначала ДНК, которую необходимо изучить, копируют, чтобы увеличить ее количество, а затем смешивают ее со специальными ДНК-микрочипами, которые помогают выявлять различные нарушения.
Эта методика позволяет в одном исследовании выявлять делеции и дупликации участков ДНК по всему геному. Разрешающая способность стандартного ХМА от 100 000 пар нуклеотидов – «букв» генетического кода (в отдельных регионах от 10 000 п. н.).
С помощью ХМА можно выявлять:
- изменения числа хромосом;
- дупликации и делеции, в том числе микродупликации и микроделеции;
- отсутствие гетерозиготности – утрату одной из двух копий гена. Это явление имеет важное значение в онкологии, при болезнях импринтинга (когда активность гена зависит от того, от какого из родителей он получен), аутосомно-рецессивных заболеваниях (связанных с рецессивными генами – о них мы поговорим ниже), близкородственных браках;
- однородительские дисомии, когда в геноме ребенка присутствуют две хромосомы от одного родителя.
Однако, как и предыдущий метод, хромосомный микроматричный анализ имеет некоторые ограничения. Он не позволяет выявлять или ограничен в выявлении таких аномалий, как:
- сбалансированные хромосомные аномалии, когда в хромосомах происходят изменения, которые не приводят к добавлению или утрате генетического материала. К ним относятся инверсии (разворот участка хромосомы на 180 градусов), реципрокные транслокации (обмен участками между хромосомами), небольшие инсерции (вставки в хромосомах);
- мозаицизм, если клеток с нарушениями в кариотипе менее 15%;
- CNV (copy number variation) – повторы небольших участков генома;
- точечные мутации – замены отдельных «букв» генетического кода;
- экспансия (увеличение) повторов коротких участков в ДНК;
- аномалии метилирования – присоединения особых метильных групп к определенным участкам ДНК, которые меняют активность генов.
Мутации в генах и заболевания, к которым они способны приводить
Мутации – это изменения, которые происходят в ДНК как случайным образом, так и под действием разных факторов, например химических веществ, ионизирующих излучений. Они могут затрагивать как отдельные «буквы» генетического кода, так и большие участки генома. Мутации происходят постоянно, и это основной двигатель эволюции. Чаще всего они бывают нейтральными, то есть ни на что не влияют, не приносят ни вреда, ни пользы. В редких случаях встречаются полезные мутации – они дают организму некоторые преимущества. Также встречаются вредные мутации – из-за них нарушается работа важных белков, наоборот, происходят достаточно часто. Генетические изменения, которые происходят более чем у 1% людей, называются полиморфизмами – это нормальная, естественная изменчивость ДНК Полиморфизмы ответственны за множество нормальных отличий между людьми, таких как цвет глаз, волос и группа крови.
Все внешние признаки и особенности работы организма, которые человек получает от родителей, передаются с помощью генов. Это важнейшее свойство всех живых организмов называется наследственностью. В зависимости от того, как проявляются гены в тех или иных признаках, их делят на две большие группы.
- Доминантные гены. Выражаясь простым языком, эти гены более «сильные». Если в клетках присутствует хотя бы одна копия такого гена, его признаки проявятся.
- Рецессивные гены «слабее» доминантных. Если у человека одна копия гена доминантная и одна рецессивная, – проявится признак доминантной. А для проявления рецессивного признака нужно две соответствующих копий.
Например, карий цвет глаз у человека является доминантным. Поэтому у кареглазых родителей с высокой вероятностью родится кареглазый ребенок. Если у одного из родителей глаза карие, а у другого голубые, то вероятность рождения кареглазых детей в такой семье тоже высока. У двух голубоглазых родителей, скорее всего, все дети тоже будут голубоглазыми. А вот у кареглазых родителей может родиться ребенок с голубыми глазами, если у обоих есть рецессивные «гены голубоглазости», и они достанутся ребенку. Конечно, это упрощенная схема, потому что за цвет глаз отвечает не один, а несколько генов, но на практике эти законы наследования зачастую работают. Аналогичным образом потомству могут передаваться и наследственные заболевания.

Как выявляют рецессивные мутации?
Для выявления мутаций, которые передаются рецессивно, используют целый ряд исследований.
Секвенирование по Сэнгеру – метод секвенирования (определения последовательности нуклеотидов, буквально – «прочтение» генетического кода) ДНК, также известен как метод обрыва цепи. Анализ используется для подтверждения выявленных мутаций. Это лучший метод для идентификации коротких тандемных повторов и секвенирования отдельных генов. Метод может обрабатывать только относительно короткие последовательности ДНК (до 300–1000 пар оснований) одновременно. Однако самым большим недостатком этого метода является большое количество времени, которое требуется для его проведения.
Если неизвестно, какую нужно выявить мутацию, то используют специальные панели.
Панель исследования — тестирование на наличие определенных мутаций, входящих в перечень конкретной панели исследования. Анализ позволяет выявить одномоментно разные мутации, которые могут приводить к генетическим заболеваниям. Анализ позволяет компоновать мутации в панели по частоте встречаемости (скрининговые панели, направленные на выявление носительства патологической мутации, часто встречаемой в данном регионе или в определенной замкнутой популяции) и по поражаемому органу или системе органов (панель «Патология соединительной ткани»). Но и у этого анализа есть ограничения. Анализ не позволяет выявить хромосомные аберрации, мозаицизм и мутации, не включенные в панель, митохондриальные заболевания, а также эпигенетические нарушения.
Не в каждой семье можно отследить все возможные рецессивные заболевания. Тогда на помощь приходит секвенирование экзома – тест для определения генетических повреждений (мутаций) в ДНК путем исследования в одном тесте практически всех областей генома, кодирующих белки, изменения которых являются причиной наследственных болезней.
Секвенирование следующего поколения-NGS – определение последовательности нуклеотидов в геномной ДНК или в совокупности информационных РНК (транскриптоме) путем амплификации (копирования) множества коротких участков генов. Это разнообразие генных фрагментов в итоге покрывает всю совокупность целевых генов или, при необходимости, весь геном.
Анализ позволяет выявить точечные мутации, вставки, делеции, инверсии и перестановки в экзоме. Анализ не позволяет выявить большие перестройки; мутации с изменением числа копий (CNV); мутации, вовлеченные в трехаллельное наследование; мутации митохондриального генома; эпигенетические эффекты; большие тринуклеотидные повторы; рецессивные мутации, связанные с Х-хромосомой, у женщин при заболеваниях, связанных с неравномерной Х-деактивацией, фенокопии и однородительские дисомии, и гены, имеющие близкие по структуре псевдогены, могут не распознаваться.

Что делать, если в семье есть наследственное заболевание?
Существуют два способа выявить наследственные генетические мутации у эмбриона:
Предимплантационное генетическое тестирование (ПГТ) в цикле ЭКО. Это диагностика генетических заболеваний у эмбриона человека перед имплантацией в слизистую оболочку матки, то есть до начала беременности. Обычно для анализа проводится биопсия одного бластомера (клетки зародыша) у эмбриона на стадии дробления (4–10 бластомеров). Существует несколько видов ПГТ: на хромосомные отклонения, на моногенные заболевания и на структурные хромосомные перестройки. Данные Simon с соавторами (2018) говорят о том, что в случае проведения ЭКО с ПГТ у пациентки 38–40 лет результативность ЭКО составляет 60%. Но при исследовании эмбриона есть ряд ограничений. Так, из-за ограниченного числа клеток можно не определить мозаицизм.
Если нет возможности провести ЭКО с ПГТ, то используют второй вариант – исследование плодного материала во время беременности.
Для забора плодного материала используют инвазивные методы:
- биопсия хориона – когда берут клетки из плаценты;
- амниоцентез – когда берут клетки амниотической жидкости.
Далее эти клетки исследуют при помощи одного или нескольких генетических тестов (которые имеют свои ограничения). Проведение инвазивных методов может быть связано с риском для беременности порядка 1%.
Таким образом, проведя дополнительные исследования, можно значительно снизить риск рождения ребенка с генетическим заболеванием в конкретной семье. Но привести этот риск к нулю на сегодняшний день, к сожалению, невозможно, так как любой генетический тест имеет ряд ограничений, что делает невозможным исключить абсолютно все генетические болезни.

Автор статьи
Пелина Ангелина Георгиевна
Врач-генетик
Ведёт генетическое обследование доноров Репробанка, осуществляет подбор доноров для пар, имеющих ранее рождённых детей с установленной генетической патологией.
+7 (499) 653-66-09
info@reprobank.ru
Записаться на консультацию
Количество выявляемых врожденных челюстно-лицевых аномалий за 40 лет выросло вдвое. По данным ВОЗ частота таких случаев составляет 1,6 на 1000 новорождённых детей, занимая второе место после пороков сердца.
 Чем опасны такие пороки
Чем опасны такие пороки
 Чем опасны такие пороки
Чем опасны такие порокиВрожденные дефекты развития лица ребенка — это одно из самых опасных осложнений беременности. В отличите от конечностей или других органов, пороки этой области даже при современном уровне челюстно лицевой хирургии устраняются плохо, приводя к инвалидности: нарушению зрения, слуха, речи, обоняния.
Лицевые дефекты часто сочетаются с пороками развития других органов или умственной отсталостью. Такие дети умирают в раннем возрасте.
Причины развития внутриутробных пороков лица
Поскольку причины врожденных аномалий детей до сих пор не изучены, ребенок с такими отклонениями может появиться практически в любой семье, вне зависимости от образа жизни, места жительства, количества родов и здоровья родителей.
Причины, существенно увеличивающие вероятность уродств:
- Неблагоприятная наследственность. Внешне здоровые родители могут быть носителями различных генных мутаций, проявляющихся у потомства. На эти отклонения приходится 7-8% врожденных патологий.
- Спонтанные мутации — нарушения процесса оплодотворения, вызванные случайными, иногда неясными причинами.
- Перенесенные во время беременности TORCH-инфекции (токсоплазмоз, краснуха, герпес, цитомегаловирусная инфекция). Неблагоприятное влияние на развитие плода оказывает вирус гриппа.
- Возраст матери старше 35 лет. В организме женщины репродуктивная функция в этот период начинает угасать, и яйцеклетки «перезревают», становясь неполноценными. Их оплодотворение чревато генетическими аномалиями.
- Сопутствующие болезни обмена веществ, патологии сердца и легких у матери. Зачатие и развитие эмбриона происходит в неблагоприятных условиях. Мешает гестации необходимость постоянного приема жизненно важных лекарств.
- Внешние факторы — проживание в экологически неблагоприятной местности, работа с вредными веществами. Некоторые тератогенные (вызывающие уродства) соединения не выводятся из организма очень долго. К ним относятся формальдегид, бензол, фенол, диоксин, мышьяк.
- Прием препаратов, влияющих на развитие ребенка. Часто уродства возникают, если женщина принимает вещества, не рекомендованные во время беременности – успокаивающие, снотворные и даже некоторые обезболивающие средства.
- Курение, употребление спиртного и наркотиков. У таких женщин риск рождения ребенка с аномалиями развития лица повышен в несколько раз.
Пациенткам, входящим в эти группы риска, необходим тщательный УЗ-контроль на протяжении всей беременности. Но поскольку патологии могут возникнуть без явных причин, такое обследование нужно пройти каждой женщине. В клинике Диана имеется современный УЗ-аппарат, позволяющий досконально рассмотреть личико малыша.
Как формируются пороки лица и неба эмбриона
У двухнедельного эмбриона на месте будущего рта уже имеется первичная ротовая ямка, которая постепенно углубляется. К концу первого месяца эта область ограничивается пятью буграми – лобным, двумя верхнечелюстными и двумя нижнечелюстными. Из них впоследствии формируется лицо и челюстной аппарат. Это процесс заканчивается примерно к седьмой неделе. Нёбо формируется к 10-11 неделе.
Неблагоприятное воздействие на плод до 11-ти недель ведет к образованию врожденных дефектов.
Аномалии развития челюстно-лицевой области видны уже на первом УЗИ скрининге, проводимом в 11-14 недель. Однако некоторые патологии выявляются при УЗ-обследованиях на более поздних сроках
Тяжелые генетические патологии, сопровождающиеся патологиями челюстно-лицевой области ребенка
| Патология | Проявление | Сопровождающие патологии | Прогноз |
| Гипертелоризм | Глаза находятся далеко от переносицы, как у животных | Сопровождает тяжелые наследственные патологии — синдромы Эдвардса, Ди Джоржи, Аперта, Нунан, Вольфа — Хиршхорна, кошачьего крика, Лойса-Дитца, Гурлера, Моркио. Дети страдают аномалиями развития органов и умственной отсталостью | Дети имеют серьезные проблемы с развитием и умирают в раннем возрасте |
| Синдром Меккеля | Выпирание мозговой ткани из полости черепа (энцефалоцеле) | Неправильное развитие почек, легких, мозга и конечностей | Дети умирают внутриутробно или в первые часы жизни |
| Краниостеноз | Ранее закрытие швов и родничков на голове ребенка. Неправильная форма черепа – плоская с одной стороны и вытянутая – с другой | Косоглазие, повышенное внутричерепное давление, судороги, эпилепсия умственная отсталость | При тяжёлой форме, регистрирующейся внутриутробно, дети остаются инвалидами |
| Голопроэнцефалия, | Отсутствие или недоразвитие носа в сочетании с близким расположением глаз и «волчьей пастью» | Мозг может быть не разделен на полушария или неправильно развит | Дети умирают в раннем возрасте или внутриутробно. |
| Микрофтальмия | Недоразвитие глаз | Слепота, судороги. У мальчиков могут наблюдаться патологии развития мочеполовой системы | Инвалидность |
| Синдром Тричера Коллинза | Неправильная форма черепа. Недоразвитие подбородка, рта и ушей | Глухота, судороги, пороки сердца | Дети часто умирают от сердечных патологий |
| Синдром Пьера- Робена | Недоразвитие нижней челюсти, расщелина неба | Катаракта, неправильное развитие органов, пороки сердца | Дети часто умирают или остаются инвалидами |
| Синдром Крузона | Неправильная форма черепа,волчья пасть, уменьшенная средняя часть лица | Нарушения зрения, судороги, пороки сердца | Может наблюдаться умственная отсталость. У больных родителей велик риск появления потомства, страдающего такой же патологией |
| Синдром Пьера Робена | Недоразвитие нижней челюсти, расщелина неба «волчья пасть» | Катаракта, слепота, патологии мочеполовой системы, сердца, неправильное развитие позвоночника, отсутствие конечностей | Дети остаются инвалидами и часто даже не могут самостоятельно передвигаться |
| Болезнь Дауна | Недоразвитие носовой кости | Пороки сердца, кривошея, неправильное развитие костей, глухота | Инвалидность из-за отклонений в умственном развитии |
| Синдром Шейтхауэра — Мари — Сентона) | Увеличение мозговой части черепа и уменьшение — лицевой | Неправильное развитие костей. Молочные зубы не сменяются постоянными до 30 лет | Интеллект не страдает, но наблюдаются проблемы с суставами и патологии ЛОР-органов |
| Синдром Шерешевского-Тёрнера | Недоразвитие нижней челюсти, низкое расположение ушей | Множественные патологии внутренних органов | Дети рано умирают от сопутствующих патологий |
| Премаксиллярная агенезия | Расщелина неба, распластанный нос, широко расставленные глаза | Недоразвитие головного мозга | Дети умирают сразу после рождения |
| Макростомия | Расщелина лица | Недоразвитие челюстей, расщелина неба, глухота, | Из-за плохого слуха и трудностей с приемом пищи возможна инвалидность |
Патологии челюстно-лицевой области, обнаруживаемые на УЗИ, которые можно устранить после рождения ребёнка
- Заячья губа – незаращение верхней губы – патология развития, устраняемая с помощью пластической операции. Не влияет на интеллект ребенка и не оставляет последствий.
- Волчья пасть – расщелина верхнего нёба. Изолированный дефект, не сопровождающемся другими патологиями, устраняют закрытием расщелины.
- Гемифациальная микросомия – недоразвитие одной половины лица. Интеллект не страдает, но ребенку понадобиться целый ряд пластических операций, по исправлению внешности.
- Косая расщелина лица – незаращение мягких тканей, идущее от угла рта к области уха. При современной технике проведения пластики дефект можно убрать, проведя несколько пластических операций.
Что делать если на УЗИ обнаружились аномалии лица или челюстей плода
Подозрение на тяжелую патологию всегда приводит родителей в шоковое состояние. Им приходится решать, стоит ли оставлять беременность.
Если у ребенка «заячья губа», записываться на аборт не стоит. В первый год жизни малыша прооперируют и он не будет отличаться от других детей. В остальных случаях, требующих проведения многоэтапных пластических операций, нужно посоветоваться с врачом-хирургом, каким будет успех таких вмешательств. Если женщина все-таки решилась сделать аборт, беременность лучше прервать на маленьком сроке медикаментозным способом.
Поскольку такие патологии могут быть как самостоятельными, так и являться симптомом тяжелых наследственных заболеваний, женщине назначаются дополнительные обследования – анализ околоплодных вод, крови из пуповины, тканей плаценты.
Если обследование показало, что ребенок родится с тяжелым наследственным недугом, родителям нужно решиться, стоит ли производить на свет больного малыша. На этот вопрос каждый отвечает сам, но прислушаться к мнению врачей в любом случае будет не лишним