
5.1 Вычисление
относительной погрешности определения
длины световой волны
.
5.1.1
Принять, что погрешность определения
значения
![]() определяется погрешностью измерений
определяется погрешностью измерений
расстоянийa
и расстояния![]() :
:
![]() (5.1)
(5.1)
где
![]() —
—
относительная погрешность определения![]() ,
,
безразмерная величина;
аср
—
среднее
значение расстояния до соответствующей
области спектра, м:
-
— фиолетовой
области в спектре I порядка — аср
кр1— фиолетовой
области в спектре II порядка — аср
кр2— красной
области в спектре I порядка — аср
ф1—
красной
области в спектре II порядка — аср
ф2
![]() —расстояние
—расстояние
от решетки до щели,м;
![]() –абсолютная
–абсолютная
погрешность в определении расстояния
![]() от решетки до щели;
от решетки до щели;
![]() –средняя
–средняя
абсолютная погрешность определения
расстояния a
от центра до соответствующей области
спектра.
Абсолютные
погрешности
![]()
и
![]() принять равными цене деления приборов:
принять равными цене деления приборов:
![]() =
=
![]() = 0,5 мм = 0,5
= 0,5 мм = 0,5![]() 10-3
10-3
м
5.1.2
Вычислить по формуле относительные
погрешности определения длины волны
![]() —
—
всего 4 значения:

— фиолетовой
области I порядка —
![]() ;
;
— фиолетовой
области II порядка —
![]() ;
;
— красной
области I порядка —
![]() ;
;
— красной
области II порядка —
![]() .
.
5.1.3
Значения
относительных погрешностей выразить
в % и записать в таблицу 3.1.
5.2 Вычисление
абсолютной погрешности определения
длины волны![]()
5.2.1
Поскольку смысл относительной погрешности:
![]() ,
,
(5.2)
Тогда:
![]() ,
,
(5.3)
где
![]() —
—
абсолютная погрешность определения
длины волны, м;
![]()
 —
—
относительная погрешность определения
длины волны (без
%) ;
![]() —
—
длина волны соответствующей области,
м:
— фиолетовой
области I порядка —
![]() ;
;
— фиолетовой
области II порядка —
![]() ;
;
— красной
области I порядка —
![]() ;
;
— красной
области II порядка —
![]() .
.
5.2.2
Вычислить по формуле (4.3) абсолютную
погрешность определения длинны волны
![]()
Всего
4 значения:
— фиолетовой
области I порядка —
![]() ;
;
— фиолетовой
области II порядка —
![]() ;
;
— красной
области I порядка —
![]() ;
;
— красной
области II порядка —
![]() .
.
5.3
Вычислить
средние значения — для спектров I и II
порядка — абсолютной и относительной
погрешностей определения длин волн
света:
—
![]() ,
,![]()
—
![]() ,
,![]()
5.4
Результаты
всех измерений записать в таблицу 3.1
5.5
Найти в
справочнике значения длин световых
волн. Записать в таблицу 3.1.
— “табличные”
значения для красного и фиолетового
света.
5.6
Полученные
экспериментально значения длин световых
волн сравнить с табличным значением
длин световых волн.
-
Запись полученных
результатов
П
 олученные
олученные
результаты – значение плотности с
абсолютной погрешностью, единицами
измерения и относительной погрешностью,
а также табличное значение записать
следующим образом:
![]() ф
ф
= (… ± …)
![]() 10-6
10-6
м,
![]() ф
ф
(табл.) = . .
. . 10— 6
м
![]() ср
ср
ф = … %


![]() кр
кр
= (… ± …)
![]() 10-6
10-6
м,
![]() кр
кр
(табл.) = . .
. . 10-6
м
![]() ср
ср
кр = … %
где
![]() ф
ф
=![]() ср
ср
ф ±
![]() ср
ср
ф ,
![]() кр
кр
=![]() ср
ср
кр±
![]() ср
ср
кр
Примечания.
-
В
окончательной записи результатов
значения длины волны красного и
фиолетового света и её абсолютной
погрешности ∆
округлить до целых значений (или до
десятых долей). -
Количество
знаков после запятой в значениях
и ∆
— должно быть одинаковым. -
Значение
относительной
погрешности
выразить в
процентах
и округлить до десятых долей процента). -
Выписать
из таблиц в справочниках или в Приложении
значение длины волны красного и
фиолетового света («табличное» значение)
—
табл. -
Не
забудьте записать единицы измерений
,
∆,
табл
!
-
Как написать
вывод о проделанной работе
Сделать
вывод с использованием рекомендаций
в таблице 7.1.
|
Помните:
Подумайте,
Таблица |
||
|
п/п |
Что должно |
Как это написать |
|
|
Что сделано в |
Напишите,
Рекомендуется В данной работе…… (Что |
|
|
Как сделано |
Краткое
|
|
|
Что получено |
Запишите |
|
|
Анализ |
Проанализируйте
Для
Например:
Напишите, |
Следует отметить, что определение длин волн по линиям сравнения, лежащим в другом порядке спектра, может привести к ошибкам, связанным с тем, что, как уже отмечалось, спектры разных порядков могут фокусироваться на несколько отстоящих друг от друга поверхностях. Если, кроме того, инструментальный контур, даваемый решеткой, асимметричен, то это может привести к кажущемуся смещению спектральных линий в спектрах разных порядков. Связанные с этими эффектами ошибки в измерении длин волн вряд ли могут превышать 0,1—0,01 А. Однако при прецизионных измерениях с погрешностями такой величины мириться нельзя. Чтобы их избежать, этот метод следует применять в сочетании с первым методом, т. е. использовать стандарты длин волн. Для этого на одну пластинку снимается неизвестный спектр элемента Л, спектр элемента В, длины волн линий которого в вакуумной области рассчитаны, и спектр элемента С, длины волн линий которого известны в видимой области и спектр первого порядка которого накладывается на линии в спектрах второго, третьего и т.д. порядков элементов Л и В. Тогда можно найти длины волн линий элемента В по линиям элемента С и сравнить их с расчетными, после чего построить кривую поправок. Это позволит для любой линии элемента Л найти поправку к измеренным длина)м волн или убедиться в том, что она пренебрежимо мала. [c.231]
Ложные полосы. При работе с кварцевыми спектрографами следует обращать специальное внимание на то, чтобы свет источника на пути к спектрографу не испытывал поляризации. Если падающий на щель спектрографа «свет поляризован, то интерференция между обыкновенными и необыкновенными лучами, на которые разделяется каждый луч в кварцевой оптике прибора, может привести к появлению в непрерывном спектре полосатой структуры, похожей на диффузные полосы эти полосы обычно столь правильны, что когда они встречаются отдельно в непрерывном спектре, их легко отличить от истинных молекулярных полос однако если они накладываются на ту или иную молекулярную систему, то могут внести ошибки в измерения длин волн и интенсивности. [c.232]
В 6. 7 рассмотрена принципиальная возможность разрешения изображений двух звезд в том случае, когда критерий Рэлея заведомо не соблюдается, но измерение суммарного контура и определение аппаратной функции могут быть проведены с малыми ошибками. Все эти рассуждения полностью применимы и к разрешению спектральным прибором двух близких по длине волны спектральных линий. [c.319]
При измерении поглощения и отражения образец лучше всего поместить за выходной щелью монохроматора, чтобы избежать возбуждения других процессов под действием излучения с длинами волн за пределами интересующей спектральной области. Например, при освещении образца с широким спектром фотоны, для которых К(й> АЕ, могут образовывать электронно-дырочные пары. Последние, рекомбинируя, будут испускать фотоны с меньшей энергией, что может привести к ошибкам в измерении поглощения. [c.167]
По схеме на фиг. 212, в компенсация достигается при помощи пластинки Х/4. Анализатор поворачивается на угол до получения темноты в рассматриваемой точке. Разность хода в числе длин волн т = (f/it. Ошибки при измерениях 1) если пластинка [c.271]
Количество звеньев в цепях между эталонами и рабочими измерительными средствами устанавливается с таким расчётом, чтобы между ошибками измерения меры или прибора и их допускаемыми погрешностями было определённое соотношение. На фиг. 118 показана схема передачи размера от эталона длины световой волны до штангенциркуля с отсчётом по нониусу в 0,05 мм. [c.223]
Из табл. 11-1 видно, что ошибка в определении истинной температуры, обусловленная неопределенностью е образца, уменьшается при смещении измерений Т я в область коротких длин волн. [c.330]
Как можно видеть из таблицы, значения Ig / для длины волны /. = = 5800 А Д)-= 10А в исследованной области температур и давлений с учетом ошибок измерения практически не меняются. Различие Big/, указанное в таблице в зависимости от температуры, лежит в пределах погрешности. Постоянство значения Ig/ в данной спектральной области с учетом ошибки измерения (порядка 0,5 в величине Ig/) означает, что интенсивность излучения могла и расти с температурой, но незначительно. [c.204]
Другим фактором, влияющим на точность измерения температуры, было то, что термометр не был защищен от действия излучения. При описании метода нагревания воды было показано, что полоса инфракрасного излучения в диапазоне длин от 0,7 до 0,97 мкм слабее всего поглощается водой, и, следовательно, эта часть энергии излучения (12% всего количества) попадает на термометр. Во всех случаях термометр находился не ближе, чем на расстоянии 2 см от стенок сосуда. Таким образом, всегда был промежуточный слой воды толщиной 2 см, поглощавший попадавшее на термометр излучение. Баллон термометра весьма прозрачен для лучей с длиной волны от 0,7 до 0,97 мкм, а ртуть в баллоне хорошо отражает и почти не поглощает излучение. Таким образом, можно сказать, что вода выполняла роль радиационной защиты термометра, благодаря чему действие радиационных эффектов на результаты измерения температуры было пренебрежимо мало по сравнению с точностью проводившихся измерений. Для проверки этого положения на термометр была надета защитная оболочка в виде алюминиевой трубки. Температуры, регистрируемые термометром без защиты, сравнивались с измерениями, проведенными с помощью защищенного термометра. Оказалось, что два одновременно замеренных значения температуры никогда не разнились больше чем на 0,1° С. Точность же термометров этого типа составляла 0,1°С. Таким образом, если учесть две наибольшие ошибки, а именно точность показаний термометра и возможности изменения температуры во время опыта, можно считать, что замеры температуры выполнялись с точностью 0,2° С. [c.242]
В широких пределах от ультрафиолетовой области и по крайней мере до 2 мк, показывают, что чувствительность термостолбика имеет максимум вблизи 1 мк и монотонно падает к 0,2 мк и к 20 мк. Типичная калибровочная кривая термостолбика приводится на фиг. 4.31. Она показывает, что если исключить систематические ошибки и погрешность измерения напряжения, то чувствительность термостолбика может быть на 13% ниже максимальной при 20 мк и на 67о ниже при 0,2 мк. Чтобы ошибка из-за спектральной зависимости не превышала 37о, с термостолбиком следует работать лишь в области длин волн от 0,2 мк до 2 мк. Блок-схема установки для измерения мощности непрерывных газовых лазеров показана на фиг. 4.32. В табл. 4.16 перечислена аппаратура, пригодная для таких измерений. [c.204]
Теоретически, конечно, стабильность (или воспроизводимость) частоты не отличается от стабильности длины волны. Но из-за отсутствия в настоящее время каких-либо эталонов частоты в области оптического спектра при нынешнем уровне развития техники частотных измерений приходится проводить одно важное различие, которое можно сопоставить с различием между случайными и систематическими ошибками в метрологии [1] ). [c.411]
Систематическая ошибка измерения не может быть меньше погрешности самого эталона, даже если случайные ошибки измерения значительно меньше. Точность оптических эталонов длины волны в настоящее время не превышает 1 Ю . Поэтому и систематическое отклонение длины волны, или абсолютная стабильность, не может быть меньше 1 10 . Таким образом, даже измерения стабильности длины волны, превышающей [c.412]
Очевидно, что расстояние между двумя соседними максимумами также равно Я/2 os 6. При уменьшении угла падения до нуля, места нулевых амплитуд обращаются в узлы, а места максимумов —в пучности стоячей волны. Это обстоятельство имеет большое значение при определении длины волны с помош ью измерения расстояния между пучностя ми или узлами в стоячей волне. Это расстояние равно Я/2 только при строгом падении луча по нормали к поверхности раздела. При отклонении угла 9 от нуля за счет неправильности установки отражателя возникает ошибка в определении длины волны, что вызывает ошибку в измерении скорости звука. Исходя из этого, в приборах — ультразвуковых интерферометрах — рефлекторы и источники плоских волн устанавливают так, чтобы угол падения был точно равен нулю. [c.185]
Следуе отметить, что при установке алюминиевой решетки 3 схеме скользящего падения коэффициент отражения меняется с длиной волны не монотонно, и это может привести к грубым ошибкам при энергетических измерениях в узкой области спектра [102] ). [c.98]
При измерении спектра мощности исследуемое излучение попадает в прибор, в котором оно подвергается разложению на спектральные компоненты и преобразуется в выходной сигнал. Окончательно этот сигнал представляется в виде зависимости от частоты или длины волны. Цель такого анализа состоит в том, чтобы по регистрируемому сигналу по возможности точно (и просто) сделать заключения о входном сигнале. Но в процессе измерений неизбежно возникают систематические ошибки, ограничивающие точность. [c.45]
В другом варианте двухволнового метода также измеряются два значения плотности на двух длинах волн первая длина волны соответствует спектральной области максимального поглощения, вторая длина волны —спектральной области прозрачности. Двухволновой метод обеспечивает ошибку измерений =ь(1- 3)%. Однако для этого необходимы хороший монохроматор, тщательная настройка оптической схемы и точное измерение коэффициентов пропускания. Так как для метода не требуется точная фокусировка изображения, то появляется возможность изучения неоднородных по толщине объектов. [c.106]
В результате прогресса лазерной техники и успешного развития радиотехнических методов преобразования частоты в оптическом диапазоне удалось существенно повысить точность измерения скорости света в вакууме. При этом проводились независимые измерения длины волн и частоты специально стаби-лизированног о неон-гелиевого лазера, генерирующего в инфракрасной области спектра (л = 3..39 мкм). Таким способом в 1972 г. скорость света была определена с большой точностью (iSf/ = 3 10 ). Авторы получили с = (299792,4562 0,0011) км/с и считают, что в дальнейшем ошибка может быть еще уменьшена за счет улучшения воспроизводимости измерения первичных эталонов длины и времени (см. 5.7). [c.51]
Вычисление ошибок проводится следующим образом. К значению одной из координат опорных линий (например, 1) добавляется величина М, равная ошибке измерений, после этого вычисляются новые ошибочные константы формулы Гартмана и находится разница между ошибочными и истинными константами АА.02, Асо2, и Аб о2. Такой расчет проводится еще два раза при изменении координат двух других опорных линий 2 и 3. По найденным разностям между ошибочными константами и истинными А 10г Асо и Ай(о1, где = 2, 3, 4, находят выборочную стандартную ошибку расчетов длины волны по основной формуле Гартмана, пользуясь формулой, полученной на основании теории ошибок [c.133]
Погрешность измерения А опреде ляется погрешностью измерения длины волны света в воздухе й ошибкой в подсчете числа интерференционных полос за четверть периода колебаний. Изменение длины волны света может быть учтено введением соответствующей поправки. Таким образом, определяющей является погрешность подсчета числа импульсов (числа интерференционных полос), на которую влияет сейсмический фон, делающий интерференционную картину неустойчивой, а также неточность установки начала и конца счета импульсов. [c.549]
Заметим в заключение, что теория относительности вообще была бы невозможна, если бы не был установлен фундаментальный факт конечности скорости распространения света. Изучение методов и результатов измерения скорости света представляет громадный, не только исторический интерес. В частности, уточнение численного значения этой постоянной необходимо для точных измерений астрономических расстояний методами радиолокации. Это в свою очередь необходимо для целей космонавтики. Однако мы не будем касаться этих вопросов. Ограничимся замечанием, что в 1972 г. скорость света была определена на основе независимых измерений длины волны X и частоты света V. Источником света служил гелий-неоновый лазер, генерировавший излучение с длиной волны 3,39 мкм. Длина волны измерялась интерферометрически сравнением ее с эталоном длины, т. е. с длиной волны в вакууме оранжевой линии изотопа криптона-86. Ошибка таких измерений 10 нм. Частота лазерного излучения измерялась путем сравнения ее с атомным стандартом частоты, т. е. с частотой перехода между двумя сверхтонкими квантовыми уровнями атома цезия-133 в нулевом магнитном поле. При этом использовались методы нелинейной оптики — генерация излучений с суммарной и разностной частотами. В итоге [c.631]
Точность измерения скорости света определяется в этом случае, во-первых, тем, насколько стабилен данный источник, и, во-вторых, тем, с какой точностью удается измерить частоту и длину волны излучения. Источниками электромагнитного излучения, наиболее удовлетворяющими этим требованиям, являются лазеры. Измерение длины В0Л1ГЫ , основанное на явлении интерференции света, производится с ошибкой, не превышающей величину порядка 10 , Измерение частоты излучения основано на технике нелинейного преобразования частоты. Используемый прибор (например, полупроводниковый диод), приняв синусоидальное колебание некоторой частоты, дает на выходе колебания более высокой частоты — удвоенной, утроенной и т. д. Этот метод с помощью нелинейного элемента излучс1П1Я кратной частоты позволяет измерять частоту излучения лазера и сравнивать его с частотами, измеренным прежде. Согласно результатам изме-рени , в1> пол 1ен ЫМ этим методом в 1972 г., скорость света в вакууме равна (299792456,2 1,1) м/с. Новые методы разработки нелинейных фотодиодов, испо.и.зусмых для смещения частот светового диапазона спектра, позволят в будущем увеличить точность лазерных измерений скорости света. [c.418]
При изучении фотографии уд шенной звезды аппаратной функцией в первом приближении является дифракционное пятно, размеры которого определяются диаметром объектива телескопа и длиной волны дифрагирующего света. Однако эта идеализированная картина существенно усложняется влиянием аберраций, полное устранение которых представляется практически невозможным. Поэтому аппаратная функция может быть определена только приближенно. Неизбежны также случайные и систематические ошибки при измерении освещенности суммарной картины. Наличие ошибок в измерении f(x — х) п Ф(х) ограничивает возможность восстановления функции объекта Дл )путем решения обратной задачи. [c.338]
Пример .6. Определить температуру плазмы по измеренному в долях интерференционного порядка значению полуширины НК Yo = 0,25 или по значению Imin = 0,23. Пусть известно, что максимальное отклонение поверхности зеркал от плоскости составляет — Х/ 0, т. е. Oi = 0,1 Яэ — = = 0,5 (i = 0,9 Дг. = 0,0017 нм Л = 0,5878) атомный вес исследуемого элемента, Ша = 4 а = as = 0. Значение yo (или /тш) измерено с ошибкой 5%. Длина световой волны X = 500 нм, толщина ИФП = 1 см. [c.151]
Необходимо заботиться о том, чтобы ошибок не вызывали интерференционные эффекты, которые часто возникают в результате многократного отражения между почти параллельными поверхностями или внутри оптических пластин. Возможность ошибки возрастает при измерениях вне видимого спектрального диапазона, ибо здесь глаз не в состоянии помочь выявить экспериментальные аномалии. Типичный пример экспериментальной ситуации, при которой возможны ошибки, — измерения мош,ности в инфракрасном диапазоне Для измерения средней мощности пользуются радиационными термостолбиками, которые мало чувствительны к длине волны (см. гл. 4). Такие термостолбики обычно содержат много термоспаев, и при их градуировке должна измеряться средняя мош,ность плоской волны. Результаты можно однозначно интерпретировать только тогда, когда измеряемый пучок однороден. Допустим, что нам нужно измерить мощность непрерывно работающего инфракрасного лазера, величина которой превышает предельную мощность, допустимую для термостолбика. Мы должны применить ослабитель, чтобы уменьшить интенсивность пучка до подходящей величины. Ослабитель можно поместить либо прямо перед термостолбиком, либо около лазера. Обычно термостолбик ставят на расстоянии 3—15 м от лазера, с тем чтобы пятно пучка равномерно освещало его апертуру. Если же ослабитель высокого качества находится около лазера, то он может образовать интерферометр Фабри — Перо и создать в пучке интерференционные полосы. Тогда термостолбик будет освещаться волновым фронтом с периодической структурой и в результате при измерениях могут возникнуть серьезные ошибки (8 1). Во избежание этого ослабитель обычно помещают около термостолбика. [c.32]
Результаты, полученные при измерениях на дифракционных спектрографах, обрабатывают другими методами. В Аргоннской лаборатории была создана установка Пашена с конфигурацией, столь близкой к круговой, что можно было измерить длины волн вполне с удовлетворительной точностью (0,001 см ), пользуясь формулой решетки тА. = d(sin 0 — sin 0 ). Лишь немногие решеточные системы подобного типа обеспечивают такую точность, так что прибор приходится калибровать по эталонам длин волн. Поскольку дисперсия нелинейна, необходимо вычислить поправочную кривую (обычно пользуются методом наименьших квад-эатов и полиномиальной аппроксимацией). При выборе эталонов необходима некоторая осторожность, так как не всегда можно сравнить линии в различных порядках (особенно для старых решеток — из-за ошибки, обусловленной затуплением резца). Поскольку более новые плоские решетки допускают такое сравнение, при выборе эталона длины волны допустима большая свобода. Почти то же самое относится к эшелле. [c.355]
На фиг. 22 изображена зависимость логарифмов светочувствительности эмульсий а, с, к я I от логарифмов их поглощательной способности. Линейная зависимость между светочувствительностью и поглощательной способностью выражалась бы прямолинейным участком прямой с наклоном 45°. Это приблизительно верно на участке между 400 и 450 т, для бромосеребряных эмульсий и между 440 и 510 для иодобромосеребря-ных эмульсий. В этом ограниченном интервале пропорциональность между светочувствительностью и поглощением подтверждается измерением обеих величин на одном и том же образце. Крутое падение кривых в области более длинных волн, лежащей вне указанных интервалов, может вызываться неактивным поглощением , которое пока не объяснено, но, весьма вероятно, обусловлено экспериментальной ошибкой в измерениях поглощения в тех случаях, когда возможна небольшая потеря света вследствие бокового рассеяния. [c.308]
Кривые оптического поглощения для чисто бромосеребряной эмульсии а показаны на фиг. 24. При —140° только короткий интервал длин волн (от 400 до 420 тр) количественно представляет поглощение фотолитически активным веществом — бромистым серебром. Вне этой области измерения поглощения значительно искажаются упомянутыми выше экспериментальными ошибками (см. п. А этого параграфа). Из фиг. 24 следует, что при 25° поглощательная способность при 420 тр равна 80% от ее значения при 400 тр и соответствующая величина при —140° составляет 55%. Это более крутое падение поглощения с длиной волны эквивалентно более крутому падению кривых на фиг. 2 (—195°) от А ==400 тр в сторону длинных волн по сравнению с кривыми, изображенными на фиг. 1 (25°), или же общему падению кривых на фиг. 3 в сторону длинных волн. Таким образом, изменение вида кривой спектральной светочувствительности между 400 и 460 тр с изменением температуры можно, повидимому, полностью объяснить изменением поглощения света. [c.312]
Вслед за первичным стандартом применяются вторичные стандарты, установленные с помощью интерфе-рометрических измерений (используется главным образом интерферометр Майкельсона). Ошибка составляет 10 нм. Вторичные и соответствующие им третичные длины волн собраны в спектральных атласах, так что в видимой области и в прилегающих к ней спектральных областях имеется большое число линий, которые могут использоваться для калибровки спектральных приборов по длинам волн. Новые успехи в этом направлении достигнуты с помощью лазерной спектроскопии высокого разрешения, особенно в инфракрасной области. [c.43]
Статические измерения парциальных давлений Pzn и Рте над ZnTe (т) различных составов были выполнены Бребриком [37 ] путем определения оптической плотности сосуществующего пара [Zn (г) + Teg (г)] как функции длины волны в ультрафиолетовой и видимой области спектра. Во всем интервале от 500 до 910° С величина Pzn над составом ZnTe, насыщенным Zn, оказалась той же, что и над чистым цинком (в пределах экспериментальной ошибки 2%), т. е. составляла 1,17 ат при 1200° К и 0,114 ат при 1000° К-Величина Рте была ниже предела чувствительности метода, т. е. <10 ат. [c.92]
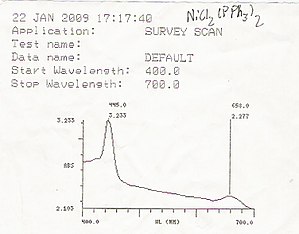
Диапазон спектроскопического анализа  УФ / видимый спектрофотометр Beckman DU640
УФ / видимый спектрофотометр Beckman DU640
Ультрафиолетовая – видимая спектроскопия или УФ-видимая спектрофотометрия (УФ-видимая или УФ / видимая ) относится к абсорбционной спектроскопии или спектроскопии отражения в части ультрафиолетовый и полный, соседний видимый спектральные области. Это означает, что он использует свет в видимом и соседнем диапазонах. Поглощение или отражение в видимом диапазоне напрямую влияет на воспринимаемый цвет используемых химических веществ. В этой области электромагнитного спектра, атомы и молекулы претерпевают электронные переходы. Абсорбционная спектроскопия дополняет флуоресцентную спектроскопию, поскольку флуоресценция имеет дело с переходами из возбужденного состояния в основное состояние, в то время как поглощение измеряет переходы из основного состояния в возбужденное.
Содержание
- 1 Принцип поглощения ультрафиолетового и видимого диапазонов
- 2 Применения
- 2.1 Практические соображения
- 2.1.1 Спектральная ширина полосы
- 2.1.2 Длина волны ошибка
- 2.1.3 Рассеянный свет
- 2.1.4 Отклонения от закона Бера – Ламберта
- 2.1.5 Источники неопределенности измерения
- 2.1 Практические соображения
- 3 Спектрофотометр ультрафиолетового и видимого диапазонов
- 4 Микроспектрофотометрия
- 5 Дополнительные приложения
- 6 См. Также
- 7 Ссылки
Принцип поглощения ультрафиолета и видимого света
Молекулы, содержащие связывающие и несвязывающие электроны (n-электроны), могут поглощать энергию в форме ультрафиолетового или видимого света. чтобы возбудить эти электроны на более высокие антисвязывающие молекулярные орбитали. Чем легче возбуждать электроны (т.е. чем меньше энергетический зазор между HOMO и LUMO ), тем большую длину волны света он может поглотить. Существует четыре возможных типа переходов (π – π *, n – π *, σ – σ * и n – σ *), и их можно упорядочить следующим образом: σ – σ *>n – σ *>π– π *>n – π *.
Применения
 Пример считывания УФ / видимой области
Пример считывания УФ / видимой области
УФ / видимая спектроскопия обычно используется в аналитической химии для количественное определение различных аналитов, таких как ионы переходных металлов, высоко конъюгированные органические соединения и биологические макромолекулы. Спектроскопический анализ обычно проводится в растворах, но также могут быть изучены твердые вещества и газы.
- Растворы ионов переходных металлов могут быть окрашены (т.е. поглощать видимый свет), потому что d-электроны внутри атомов металла могут быть возбуждены из одного электронного состояния в другое. На цвет растворов ионов металлов сильно влияет присутствие других частиц, таких как определенные анионы или лиганды. Например, разбавленный раствор сульфата меди имеет очень светло-голубой цвет; добавление аммиака усиливает цвет и изменяет длину волны максимального поглощения (λ max).
- Органические соединения, особенно с высокой степенью конъюгации, также поглощают свет в УФ или видимой областях электромагнитного спектра. Растворителями для этих определений часто являются вода для водорастворимых соединений или этанол для растворимых в органических веществах соединений (органические растворители могут обладают значительным УФ-поглощением; не все растворители подходят для использования в УФ-спектроскопии. Этанол очень слабо поглощает на большинстве длин волн.) Полярность растворителя и pH могут влиять на спектр поглощения органического соединения. Тирозин, например, увеличивает максимумы поглощения и молярную долю коэффициент экстинкции при увеличении pH от 6 до 13 или при уменьшении полярности растворителя.
- Хотя комплексы с переносом заряда также вызывают появление цветов, цвета часто слишком интенсивны, чтобы их можно было использовать для количественного измерения.
Закон Бера – Ламберта гласит: t что поглощение раствора прямо пропорционально концентрации поглощающих веществ в растворе и длине пути. Таким образом, для фиксированной длины пути УФ / видимая спектроскопия может использоваться для определения концентрации поглотителя в растворе. Необходимо знать, насколько быстро изменяется поглощение при концентрации. Это может быть взято из справочных материалов (таблицы молярных коэффициентов экстинкции ) или, более точно, определено по калибровочной кривой.
. Спектрофотометр УФ / видимой области может использоваться в качестве детектора для ВЭЖХ. Присутствие аналита дает ответ, который, как предполагается, пропорционален концентрации. Для получения точных результатов реакцию прибора на неизвестный аналит следует сравнивать с ответом на стандарт; это очень похоже на использование калибровочных кривых. Отклик (например, высота пика) для конкретной концентрации известен как коэффициент отклика.
Длины волн пиков поглощения могут быть коррелированы с типами связей в данной молекуле и важны для определения функциональных групп внутри молекула. Например, правила Вудворда – Физера представляют собой набор эмпирических наблюдений, используемых для прогнозирования λ max, длины волны наиболее интенсивного поглощения УФ / видимого света для сопряженных органических соединений, таких как диены и кетоны. Однако сам по себе спектр не является специфическим тестом для какого-либо конкретного образца. Природа растворителя, pH раствора, температура, высокие концентрации электролита и присутствие мешающих веществ могут влиять на спектр поглощения. Экспериментальные вариации, такие как ширина щели (эффективная полоса пропускания) спектрофотометра, также изменят спектр. Чтобы применить УФ / видимую спектроскопию к анализу, эти переменные должны контролироваться или учитываться, чтобы идентифицировать присутствующие вещества.
Метод чаще всего используется в количественном отношении для определения концентраций поглощающих веществ в растворе., используя закон Бера – Ламберта :
- A = log 10 (I 0 / I) = ε c L { displaystyle A = log _ {10} (I_ {0} / I) = varepsilon cL}
 ,
,
 ,
,где A — измеренное поглощение (в единицах поглощения (AU)), I 0 { displaystyle I_ {0}} — интенсивность падающий свет с заданной длиной волны , I { displaystyle I}
— интенсивность падающий свет с заданной длиной волны , I { displaystyle I} — это передаваемая интенсивность, L — длина пути через образец, c — концентрация поглощающего вида. Для каждого вида и длины волны ε является постоянной величиной, известной как молярная поглощающая способность или коэффициент экстинкции. Эта константа является фундаментальным молекулярным свойством в данном растворителе, при определенной температуре и давлении и имеет единицы 1 / M * см { displaystyle 1 / M * cm}
— это передаваемая интенсивность, L — длина пути через образец, c — концентрация поглощающего вида. Для каждого вида и длины волны ε является постоянной величиной, известной как молярная поглощающая способность или коэффициент экстинкции. Эта константа является фундаментальным молекулярным свойством в данном растворителе, при определенной температуре и давлении и имеет единицы 1 / M * см { displaystyle 1 / M * cm} .
.
Поглощение и экстинкция ε иногда определяется в терминах натурального логарифма вместо десятичного логарифма.
Закон Бера – Ламберта полезен для характеристики многих соединений, но не является универсальным соотношением для концентрации и поглощения всех веществ. Полиномиальное соотношение 2-го порядка между абсорбцией и концентрацией иногда встречается для очень больших сложных молекул, таких как органические красители (например, ксиленоловый оранжевый или нейтральный красный )..
УФ-видимая спектроскопия также используется в полупроводниковой промышленности для измерения толщины и оптических свойств тонких пленок на пластине. УФ-видимые спектрометры используются для измерения коэффициента отражения света и могут быть проанализированы с помощью дисперсионных уравнений Форухи – Блумера для определения показателя преломления (n) и коэффициента экстинкции (k) данной пленки. во всем измеренном спектральном диапазоне.
Практические соображения
Закон Бера – Ламберта содержит неявные предположения, которые должны быть выполнены экспериментально, чтобы его можно было применить; в противном случае возможны отклонения от закона. Например, химический состав и физическая среда образца могут изменить его коэффициент экстинкции. Поэтому химические и физические условия испытуемого образца должны соответствовать эталонным измерениям, чтобы выводы были достоверными. Во всем мире фармакопеи, такие как Американская (USP) и Европейская (Ph. Eur.) Фармакопеи, требуют, чтобы спектрофотометры работали в соответствии со строгими нормативными требованиями, включая такие факторы, как рассеянный свет и точность длины волны.
Спектральная полоса пропускания
Важно иметь источник монохроматического излучения для света, падающего на ячейку с образцом. Монохроматичность измеряется как ширина «треугольника», образованного всплеском интенсивности, равная половине максимальной интенсивности. Данный спектрометр имеет спектральную полосу пропускания, которая характеризует, насколько монохроматическим падающий свет. Если эта ширина полосы сравнима (или больше) с шириной линии поглощения, то измеренный коэффициент экстинкции будет ошибочным. При эталонных измерениях ширина полосы частот прибора (ширина полосы падающего света) остается ниже ширины спектральных линий. Когда исследуемый материал измеряется, ширина полосы падающего света также должна быть достаточно узкой. Уменьшение спектральной полосы пропускания уменьшает энергию, передаваемую на детектор, и, следовательно, потребует более длительного времени измерения для достижения того же отношения сигнал / шум.
Погрешность длины волны
В жидкостях коэффициент экстинкции обычно медленно изменяется с длиной волны. Пик кривой поглощения (длина волны, при которой поглощение достигает максимума) — это место, где скорость изменения поглощения с длиной волны наименьшая. Измерения обычно производятся на пике, чтобы минимизировать ошибки, вызванные ошибками в длине волны в приборе, то есть ошибками из-за того, что коэффициент ослабления отличается от предполагаемого.
Рассеянный свет
Еще одним важным фактором является чистота используемого света. Наиболее важным фактором, влияющим на это, является уровень рассеянного света монохроматора..
Используемый детектор является широкополосным; он реагирует на весь свет, который его достигает. Если значительное количество света, прошедшего через образец, содержит длины волн, которые имеют гораздо более низкие коэффициенты экстинкции, чем номинальный, прибор сообщит о неверно низком поглощении. Любой прибор достигнет точки, когда увеличение концентрации образца не приведет к увеличению зарегистрированного поглощения, потому что детектор просто реагирует на рассеянный свет. На практике концентрацию образца или длину оптического пути необходимо отрегулировать так, чтобы неизвестная оптическая плотность находилась в пределах диапазона, допустимого для прибора. Иногда эмпирическая калибровочная функция разрабатывается с использованием известных концентраций образца, чтобы позволить измерения в той области, где прибор становится нелинейным.
В качестве приблизительного примера можно сказать, что прибор с одним монохроматором обычно имеет уровень паразитного света, соответствующий примерно 3 единицам поглощения (AU), что делает измерения выше примерно 2 AU проблематичными. Более сложный прибор с двойным монохроматором будет иметь уровень паразитного света, соответствующий примерно 6 а.е., что, следовательно, позволит измерять гораздо более широкий диапазон поглощения.
Отклонения от закона Бера – Ламберта
При достаточно высоких концентрациях полосы поглощения насыщаются и демонстрируют выравнивание поглощения. Пик поглощения кажется сглаженным, потому что почти 100% света уже поглощается. Концентрация, при которой это происходит, зависит от конкретного измеряемого соединения. Один из тестов, который можно использовать для проверки этого эффекта, — это изменение длины пути измерения. В законе Бера-Ламберта изменение концентрации и длины пути имеет эквивалентный эффект — разбавление раствора в 10 раз дает тот же эффект, что и сокращение длины пути в 10 раз. Если доступны ячейки с разной длиной пути, тестирование если это соотношение верно, это один из способов судить, происходит ли выравнивание поглощения.
Неоднородные растворы могут отличаться от закона Бера – Ламберта из-за явления сглаживания поглощения. Это может произойти, например, когда поглощающее вещество находится внутри взвешенных частиц. Отклонения будут наиболее заметны в условиях низкой концентрации и высокого поглощения. В последней ссылке описан способ исправления этого отклонения.
Некоторые растворы, такие как хлорид меди (II) в воде, визуально изменяются при определенной концентрации из-за изменения условий вокруг окрашенного иона (иона двухвалентной меди). Для хлорида меди (II) это означает сдвиг от синего к зеленому, что будет означать, что монохроматические измерения будут отклоняться от закона Бера-Ламберта.
Источники неопределенности измерения
Вышеуказанные факторы влияют на неопределенность измерения результатов, полученных с помощью спектрофотометрии в УФ / видимом диапазоне. Если УФ / видимая спектрофотометрия используется в количественном химическом анализе, то на результаты дополнительно влияют источники неопределенности, возникающие из-за природы измеряемых соединений и / или растворов. К ним относятся спектральные помехи, вызванные перекрытием полос поглощения, выцветанием цвета поглощающих частиц (вызванным разложением или реакцией) и возможным несоответствием состава между образцом и калибровочным раствором.
Ультрафиолетовый – видимый спектрофотометр
Прибор , используемый в ультрафиолетовой и видимой спектроскопии, называется спектрофотометром UV / Vis . Он измеряет интенсивность света после прохождения через образец (I { displaystyle I}) и сравнивает ее с интенсивностью света до того, как он пройдет через образец (I o { Displaystyle I_ {o}} ). Отношение I / I o { displaystyle I / I_ {o}}
). Отношение I / I o { displaystyle I / I_ {o}} называется коэффициентом пропускания и обычно выражается в процентах (% T). поглощение, A { displaystyle A}
называется коэффициентом пропускания и обычно выражается в процентах (% T). поглощение, A { displaystyle A} основано на коэффициенте пропускания:
основано на коэффициенте пропускания:
- A = — log (% T / 100%) { displaystyle A = — log (% T / 100 %)}

Спектрофотометр УФ – видимого диапазонов также можно настроить для измерения коэффициента отражения. В этом случае спектрофотометр измеряет интенсивность света, отраженного от образца (I { displaystyle I}), и сравнивает ее с интенсивностью света, отраженного от эталонного материала (I o { displaystyle I_ {o}}) (например, белая плитка). Отношение I / I o { displaystyle I / I_ {o}}называется отражательной способностью и обычно выражается в процентах (% R).
Основными частями спектрофотометра являются источник света, держатель для образца, дифракционная решетка в монохроматоре или призма для разделения разных длин волн света и детектора. Источником излучения часто является вольфрамовая нить накала (300–2500 нм), дейтериевая дуговая лампа, непрерывная в ультрафиолетовой области (190–400 нм), ксенон. дуговая лампа, непрерывная от 160 до 2000 нм; или в последнее время светоизлучающие диоды (LED) для видимых длин волн. Детектор обычно представляет собой фотоэлектронный умножитель , фотодиод, матрицу фотодиодов или устройство с зарядовой связью (CCD). Детекторы с одним фотодиодом и фотоэлектронные умножители используются со сканирующими монохроматорами, которые фильтруют свет так, что только свет одной длины волны достигает детектора за один раз. Сканирующий монохроматор перемещает дифракционную решетку для «сквозного» перехода на каждую длину волны, так что ее интенсивность может быть измерена как функция длины волны. Фиксированные монохроматоры используются с ПЗС-матрицами и матрицами фотодиодов. Поскольку оба этих устройства состоят из множества детекторов, сгруппированных в одно- или двухмерные массивы, они могут собирать свет с разной длиной волны на разных пикселях или группах пикселей одновременно.
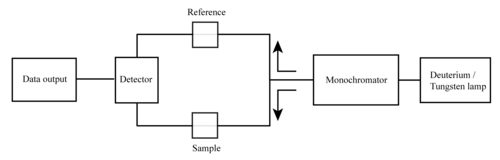 Упрощенная схема двухлучевого УФ – видимого спектрофотометра
Упрощенная схема двухлучевого УФ – видимого спектрофотометра
Спектрофотометр может быть однолучевым или двухлучевым. В однолучевом приборе (таком как Spectronic 20 ) весь свет проходит через ячейку для образца. I o { displaystyle I_ {o}}должен быть измерен путем удаления образца. Это был самый ранний дизайн, который до сих пор широко используется как в учебных, так и в промышленных лабораториях.
В двухлучевом приборе свет разделяется на два луча, прежде чем достигнет образца. Один луч используется как эталонный; другой луч проходит через образец. За эталонную интенсивность луча принимается 100% пропускание (или 0 абсорбция), а отображаемое измерение представляет собой отношение двух интенсивностей луча. Некоторые двухлучевые приборы имеют два детектора (фотодиоды), и образец и эталонный луч измеряются одновременно. В других приборах два луча проходят через прерыватель луча , который блокирует один луч за раз. Детектор попеременно измеряет образец пучка и опорный пучок синхронно с прерывателем. В цикле измельчения также может быть один или несколько темных интервалов. В этом случае измеренные интенсивности луча могут быть скорректированы путем вычитания интенсивности, измеренной в темном интервале, прежде чем будет принято соотношение.
В однолучевом приборе сначала необходимо измерить кювету, содержащую только растворитель. Компания Mettler Toledo разработала однолучевой спектрофотометр, который позволяет проводить быстрые и точные измерения в УФ / видимом диапазоне. Источник света состоит из ксеноновой лампы-вспышки для ультрафиолетового (УФ), а также для видимого (VIS) и ближнего инфракрасного диапазона длин волн, охватывающего спектральный диапазон от 190 до 1100 нм. Вспышки лампы фокусируются на стекловолокне, которое направляет луч света на кювету, содержащую раствор образца. Луч проходит через образец, и его компоненты поглощают волны определенной длины. Оставшийся свет собирается после кюветы с помощью стекловолокна и подается в спектрограф. Спектрограф состоит из дифракционной решетки, которая разделяет свет на разные длины волн, и ПЗС-датчика для записи данных соответственно. Таким образом, весь спектр измеряется одновременно, что обеспечивает быструю регистрацию.
Образцы для спектрофотометрии в УФ / видимом диапазоне чаще всего представляют собой жидкости, хотя также можно измерить поглощение газов и даже твердых веществ. Образцы обычно помещаются в прозрачную ячейку , известную как кювета. Кюветы обычно имеют прямоугольную форму, обычно с внутренней шириной 1 см. (Эта ширина становится длиной пути, L { displaystyle L} в законе Бера – Ламберта.) Пробирки также могут использоваться в качестве кювет в некоторых приборах. Тип используемого контейнера для образца должен позволять излучению проходить через интересующую спектральную область. Наиболее широко применяемые кюветы изготовлены из высококачественного плавленого кварца или кварцевого стекла, поскольку они прозрачны в УФ, видимой и ближней инфракрасной областях. Стеклянные и пластмассовые кюветы также широко распространены, хотя стекло и большинство пластмасс поглощают УФ-излучение, что ограничивает их применимость к видимым длинам волн.
в законе Бера – Ламберта.) Пробирки также могут использоваться в качестве кювет в некоторых приборах. Тип используемого контейнера для образца должен позволять излучению проходить через интересующую спектральную область. Наиболее широко применяемые кюветы изготовлены из высококачественного плавленого кварца или кварцевого стекла, поскольку они прозрачны в УФ, видимой и ближней инфракрасной областях. Стеклянные и пластмассовые кюветы также широко распространены, хотя стекло и большинство пластмасс поглощают УФ-излучение, что ограничивает их применимость к видимым длинам волн.
Также были изготовлены специальные инструменты. К ним относятся прикрепление спектрофотометров к телескопам для измерения спектров астрономических объектов. Микроспектрофотометры УФ – видимого диапазона состоят из микроскопа УФ – видимого света , интегрированного со спектрофотометром УФ – видимого диапазона.
Полный спектр поглощения на всех интересующих длинах волн часто может быть получен непосредственно с помощью более сложного спектрофотометра. В более простых приборах поглощение определяется по длине волны за раз, а затем оператором составляется спектр. Удалив зависимость от концентрации, можно определить коэффициент экстинкции (ε) как функцию длины волны.
Микроспектрофотометрия
УФ – видимая спектроскопия микроскопических образцов выполняется путем интеграции оптического микроскопа с оптикой УФ – видимого диапазона, источниками белого света, монохроматором и чувствительным детектором например, устройство с зарядовой связью (CCD) или фотоумножитель трубка (PMT). Поскольку доступен только один оптический путь, это однолучевые приборы. Современные приборы способны измерять УФ-видимые спектры как по отражательной способности, так и по пропусканию в областях отбора проб микронного масштаба. Преимущества использования таких инструментов заключаются в том, что они могут измерять микроскопические образцы, но также могут измерять спектры более крупных образцов с высоким пространственным разрешением. Таким образом, они используются в судебно-медицинской лаборатории для анализа красителей и пигментов в отдельных текстильных волокнах, микроскопических осколков краски и цвета осколков стекла. Они также используются в материаловедении и биологических исследованиях, а также для определения содержания энергии в угле и нефтематеринских породах путем измерения отражательной способности витринита. Микроспектрофотометры используются в полупроводниковой и микрооптической промышленности для контроля толщины тонких пленок после их нанесения. В полупроводниковой промышленности они используются, потому что критические размеры схемы микроскопические. Типичное испытание полупроводниковой пластины включает в себя получение спектров из многих точек на пластине с рисунком или без него. Толщина осажденных пленок может быть вычислена по интерференционной картине спектров. Кроме того, спектрофотометрия в ультрафиолетовом и видимом диапазонах может использоваться для определения толщины, а также показателя преломления и коэффициента экстинкции тонких пленок, как описано в Показатель преломления и коэффициент экстинкции тонкопленочных материалов. Затем можно создать карту толщины пленки по всей пластине и использовать ее для контроля качества.
Дополнительные приложения
УФ / видимый свет можно применять для определения кинетики или константы скорости химическая реакция. Реакция, протекающая в растворе, должна приводить к изменению цвета или яркости от реагентов к продуктам, чтобы использовать УФ / видимый свет для этого применения. Например, молекула дитизоната ртути имеет желто-оранжевый цвет в разбавленном растворе (1 * 10 ^ -5 M) и становится синим при воздействии определенных длин волн видимого света (и УФ) через конформационное изменение, но эта реакция обратимо обратно в желтое «основное состояние».
Используя оптические волокна в качестве передающего элемента спектра горючих газов, можно определить химический состав топлива, температуру газов и соотношение воздух-топливо.
Константу скорости конкретной реакции можно определить путем измерения спектра поглощения УФ / видимой области через определенные интервалы времени. Снова используя дитизонат ртути в качестве примера, можно направить свет на образец, чтобы раствор стал синим, а затем запускать УФ / видимый тест каждые 10 секунд (переменная), чтобы увидеть, как уровни поглощенной и отраженной длин волн меняются с течением времени в соответствии с раствор снова становится желтым из возбужденного синего энергетического состояния. По этим измерениям можно рассчитать концентрацию двух видов. Реакция дитизоната ртути от одной конформации к другой является реакцией первого порядка и будет иметь интегральный закон скорости первого порядка: ln [A] (время t) = — kt + ln [A] (начальное). Следовательно, построение графика натурального логарифма (ln) концентрации [A] в зависимости от времени приведет к графику линии с наклоном -k или отрицательной константой скорости. Различные порядки скорости имеют разные интегрированные законы скорости в зависимости от механизма реакции.
Константу равновесия также можно рассчитать с помощью УФ / видимой спектроскопии. После определения оптимальных длин волн для всех частиц, участвующих в равновесии, реакцию можно запустить до равновесия, и концентрацию компонентов определить с помощью спектроскопии на различных известных длинах волн. Константу равновесия можно рассчитать как K (экв) = [Продукты] / [Реагенты].
См. Также
Литература
10.6 Определение погрешности установки длины волны
10.6.1 Погрешность при установке длины волны определяют с использованием ртутной лампы (ламп), эмиссионный пик (пики) которой является референтным. Если в спектрофотометре невозможна установка лампы, то используют референтные фильтры с известными линиями поглощения, например гольмиевый. Спектр поглощения должен быть измерен с шагом <= 0,1 нм. Выбирают три различных пика для тестирования во всем диапазоне длин волн. Длину волны каждого из выбранных пиков измеряют на спектрофотометре n раз.
10.6.2 Среднее значение ![]() по результатам измерений длины волны для каждого пика рассчитывают по формуле
по результатам измерений длины волны для каждого пика рассчитывают по формуле
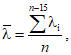 (4)
(4)
где ![]() — измеренное значение длины волны, нм;
— измеренное значение длины волны, нм;
n — число измерений, равное 15.
10.6.3 Значение абсолютной случайной составляющей погрешности S (СКО) измерения длины волны рассчитывают по формуле
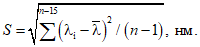 (5)
(5)
10.6.4 Спектрофотометр признают прошедшим поверку, если выполняются неравенства
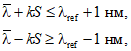 (6)
(6)
где k = 2,95 при доверительной вероятности p = 0,95 и n = 15;
![]() — значение длины волны светофильтра, приведенное в свидетельстве о поверке или эмиссионного пика ртутной лампы.
— значение длины волны светофильтра, приведенное в свидетельстве о поверке или эмиссионного пика ртутной лампы.
Значение абсолютной погрешности установки длины волны не должно превышать 1 нм.
Этот способ определения погрешности установки длины волны не может быть точным для спектрофотометров, источником излучения в которых являются лампы с линейным спектром.
В случае использования интерференционных фильтров в спектрофотометре определяют спектральную ширину используемого интерференционного фильтра на половине высоты пика пропускания с помощью спектрометра с высоким разрешением, для чего интерференционный фильтр извлекают из спектрофотометра.
Скачать документ целиком в формате PDF
Beckman DU640 UV/Vis spectrophotometer
UV spectroscopy or UV–visible spectrophotometry (UV–Vis or UV/Vis) refers to absorption spectroscopy or reflectance spectroscopy in part of the ultraviolet and the full, adjacent visible regions of the electromagnetic spectrum.[1] Being relatively inexpensive and easily implemented, this methodology is widely used in diverse applied and fundamental applications. The only requirement is that the sample absorb in the UV-Vis region, i.e. be a chromophore. Absorption spectroscopy is complementary to fluorescence spectroscopy. Parameters of interest, besides the wavelength of measurement, are absorbance (A) or transmittance (%T) or reflectance (%R), and its change with time.[2][3]
Optical transitions[edit]
Most molecules and ions absorb energy in the ultraviolet or visible range, i.e., they are chromophores. The absorbed photon excites an electron in the chromophore to higher energy molecular orbitals, giving rise to an excited state.[4] For organic chromophores, four possible types of transitions are assumed: π–π*, n–π*, σ–σ*, and n–σ*. Transition metal complexes are often colored (i.e., absorb visible light) owing to the presence of multiple electronic states associated with incompletely filled d orbitals.[3]
Applications[edit]
An example of a UV/Vis readout
UV/Vis can be used to monitor structural changes in DNA.[1]
UV/Vis spectroscopy is routinely used in analytical chemistry for the quantitative determination of diverse analytes or sample, such as transition metal ions, highly conjugated organic compounds, and biological macromolecules. Spectroscopic analysis is commonly carried out in solutions but solids and gases may also be studied.
- Organic compounds, especially those with a high degree of conjugation, also absorb light in the UV or visible regions of the electromagnetic spectrum. The solvents for these determinations are often water for water-soluble compounds, or ethanol for organic-soluble compounds. (Organic solvents may have significant UV absorption; not all solvents are suitable for use in UV spectroscopy. Ethanol absorbs very weakly at most wavelengths.) Solvent polarity and pH can affect the absorption spectrum of an organic compound. Tyrosine, for example, increases in absorption maxima and molar extinction coefficient when pH increases from 6 to 13 or when solvent polarity decreases.
- While charge transfer complexes also give rise to colours, the colours are often too intense to be used for quantitative measurement.
The Beer–Lambert law states that the absorbance of a solution is directly proportional to the concentration of the absorbing species in the solution and the path length.[5] Thus, for a fixed path length, UV/Vis spectroscopy can be used to determine the concentration of the absorber in a solution. It is necessary to know how quickly the absorbance changes with concentration. This can be taken from references (tables of molar extinction coefficients), or more accurately, determined from a calibration curve.
A UV/Vis spectrophotometer may be used as a detector for HPLC. The presence of an analyte gives a response assumed to be proportional to the concentration. For accurate results, the instrument’s response to the analyte in the unknown should be compared with the response to a standard; this is very similar to the use of calibration curves. The response (e.g., peak height) for a particular concentration is known as the response factor.
The wavelengths of absorption peaks can be correlated with the types of bonds in a given molecule and are valuable in determining the functional groups within a molecule. The Woodward–Fieser rules, for instance, are a set of empirical observations used to predict λmax, the wavelength of the most intense UV/Vis absorption, for conjugated organic compounds such as dienes and ketones. The spectrum alone is not, however, a specific test for any given sample. The nature of the solvent, the pH of the solution, temperature, high electrolyte concentrations, and the presence of interfering substances can influence the absorption spectrum. Experimental variations such as the slit width (effective bandwidth) of the spectrophotometer will also alter the spectrum. To apply UV/Vis spectroscopy to analysis, these variables must be controlled or accounted for in order to identify the substances present.[6]
The method is most often used in a quantitative way to determine concentrations of an absorbing species in solution, using the Beer–Lambert law:
- ,
where A is the measured absorbance (formally dimensionless but generally reported in Absorbance Units (AU)[7]), is the intensity of the incident light at a given wavelength, is the transmitted intensity, L the path length through the sample, and c the concentration of the absorbing species. For each species and wavelength, ε is a constant known as the molar absorptivity or extinction coefficient. This constant is a fundamental molecular property in a given solvent, at a particular temperature and pressure, and has units of .
The absorbance and extinction ε are sometimes defined in terms of the natural logarithm instead of the base-10 logarithm.
The Beer–Lambert Law is useful for characterizing many compounds but does not hold as a universal relationship for the concentration and absorption of all substances. A 2nd order polynomial relationship between absorption and concentration is sometimes encountered for very large, complex molecules such as organic dyes (Xylenol Orange or Neutral Red, for example).[citation needed]
UV–Vis spectroscopy is also used in the semiconductor industry to measure the thickness and optical properties of thin films on a wafer. UV–Vis spectrometers are used to measure the reflectance of light, and can be analyzed via the Forouhi–Bloomer dispersion equations to determine the index of refraction ( ) and the extinction coefficient (
) and the extinction coefficient ( ) of a given film across the measured spectral range.[8]
) of a given film across the measured spectral range.[8]
Practical considerations[edit]
The Beer–Lambert law has implicit assumptions that must be met experimentally for it to apply; otherwise there is a possibility of deviations from the law.[9] For instance, the chemical makeup and physical environment of the sample can alter its extinction coefficient. The chemical and physical conditions of a test sample therefore must match reference measurements for conclusions to be valid. Worldwide, pharmacopoeias such as the American (USP) and European (Ph. Eur.) pharmacopeias demand that spectrophotometers perform according to strict regulatory requirements encompassing factors such as stray light[10] and wavelength accuracy.[11]
Spectral bandwidth[edit]
It is important to have a monochromatic source of radiation for the light incident on the sample cell.[9] Monochromaticity is measured as the width of the «triangle» formed by the intensity spike, at one half of the peak intensity. A given spectrometer has a spectral bandwidth that characterizes how monochromatic the incident light is.[clarification needed] If this bandwidth is comparable to (or more than) the width of the absorption line, then the measured extinction coefficient will be mistaken. In reference measurements, the instrument bandwidth (bandwidth of the incident light) is kept below the width of the spectral lines. When a test material is being measured, the bandwidth of the incident light should also be sufficiently narrow. Reducing the spectral bandwidth reduces the energy passed to the detector and will, therefore, require a longer measurement time to achieve the same signal to noise ratio.
Wavelength error[edit]
In liquids, the extinction coefficient usually changes slowly with wavelength. A peak of the absorbance curve (a wavelength where the absorbance reaches a maximum) is where the rate of change in absorbance with wavelength is smallest.[9] Measurements are usually made at a peak to minimize errors produced by errors in wavelength in the instrument, that is errors due to having a different extinction coefficient than assumed.
Stray light[edit]
Another important major factor is the purity of the light used. The most important factor affecting this is the stray light level of the monochromator.[9]
The detector used is broadband; it responds to all the light that reaches it. If a significant amount of the light passed through the sample contains wavelengths that have much lower extinction coefficients than the nominal one, the instrument will report an incorrectly low absorbance. Any instrument will reach a point where an increase in sample concentration will not result in an increase in the reported absorbance, because the detector is simply responding to the stray light. In practice the concentration of the sample or the optical path length must be adjusted to place the unknown absorbance within a range that is valid for the instrument. Sometimes an empirical calibration function is developed, using known concentrations of the sample, to allow measurements into the region where the instrument is becoming non-linear.
As a rough guide, an instrument with a single monochromator would typically have a stray light level corresponding to about 3 Absorbance Units (AU), which would make measurements above about 2 AU problematic. A more complex instrument with a double monochromator would have a stray light level corresponding to about 6 AU, which would therefore allow measuring a much wider absorbance range.
Deviations from the Beer–Lambert law[edit]
At sufficiently high concentrations, the absorption bands will saturate and show absorption flattening. The absorption peak appears to flatten because close to 100% of the light is already being absorbed. The concentration at which this occurs depends on the particular compound being measured. One test that can be used to test for this effect is to vary the path length of the measurement. In the Beer–Lambert law, varying concentration and path length has an equivalent effect—diluting a solution by a factor of 10 has the same effect as shortening the path length by a factor of 10. If cells of different path lengths are available, testing if this relationship holds true is one way to judge if absorption flattening is occurring.
Solutions that are not homogeneous can show deviations from the Beer–Lambert law because of the phenomenon of absorption flattening. This can happen, for instance, where the absorbing substance is located within suspended particles.[12][13] The deviations will be most noticeable under conditions of low concentration and high absorbance. The last reference describes a way to correct for this deviation.
Some solutions, like copper(II)chloride in water, change visually at a certain concentration because of changed conditions around the coloured ion (the divalent copper ion). For copper(II)chloride it means a shift from blue to green,[14] which would mean that monochromatic measurements would deviate from the Beer–Lambert law.
Measurement uncertainty sources[edit]
The above factors contribute to the measurement uncertainty of the results obtained with UV/Vis spectrophotometry. If UV/Vis spectrophotometry is used in quantitative chemical analysis then the results are additionally affected by uncertainty sources arising from the nature of the compounds and/or solutions that are measured. These include spectral interferences caused by absorption band overlap, fading of the color of the absorbing species (caused by decomposition or reaction) and possible composition mismatch between the sample and the calibration solution.[15]
Ultraviolet–visible spectrophotometer[edit]
The instrument used in ultraviolet–visible spectroscopy is called a UV/Vis spectrophotometer. It measures the intensity of light after passing through a sample (), and compares it to the intensity of light before it passes through the sample (). The ratio is called the transmittance, and is usually expressed as a percentage (%T). The absorbance, , is based on the transmittance:
The UV–visible spectrophotometer can also be configured to measure reflectance. In this case, the spectrophotometer measures the intensity of light reflected from a sample (), and compares it to the intensity of light reflected from a reference material () (such as a white tile). The ratio is called the reflectance, and is usually expressed as a percentage (%R).
The basic parts of a spectrophotometer are a light source, a holder for the sample, a diffraction grating in a monochromator or a prism to separate the different wavelengths of light, and a detector. The radiation source is often a Tungsten filament (300–2500 nm), a deuterium arc lamp, which is continuous over the ultraviolet region (190–400 nm), Xenon arc lamp, which is continuous from 160 to 2,000 nm; or more recently, light emitting diodes (LED)[2] for the visible wavelengths. The detector is typically a photomultiplier tube, a photodiode, a photodiode array or a charge-coupled device (CCD). Single photodiode detectors and photomultiplier tubes are used with scanning monochromators, which filter the light so that only light of a single wavelength reaches the detector at one time. The scanning monochromator moves the diffraction grating to «step-through» each wavelength so that its intensity may be measured as a function of wavelength. Fixed monochromators are used with CCDs and photodiode arrays. As both of these devices consist of many detectors grouped into one or two dimensional arrays, they are able to collect light of different wavelengths on different pixels or groups of pixels simultaneously.
Simplified schematic of a double beam UV–visible spectrophotometer
A spectrophotometer can be either single beam or double beam. In a single beam instrument (such as the Spectronic 20), all of the light passes through the sample cell. must be measured by removing the sample. This was the earliest design and is still in common use in both teaching and industrial labs.
In a double-beam instrument, the light is split into two beams before it reaches the sample. One beam is used as the reference; the other beam passes through the sample. The reference beam intensity is taken as 100% Transmission (or 0 Absorbance), and the measurement displayed is the ratio of the two beam intensities. Some double-beam instruments have two detectors (photodiodes), and the sample and reference beam are measured at the same time. In other instruments, the two beams pass through a beam chopper, which blocks one beam at a time. The detector alternates between measuring the sample beam and the reference beam in synchronism with the chopper. There may also be one or more dark intervals in the chopper cycle. In this case, the measured beam intensities may be corrected by subtracting the intensity measured in the dark interval before the ratio is taken.
In a single-beam instrument, the cuvette containing only a solvent has to be measured first. Mettler Toledo developed a single beam array spectrophotometer that allows fast and accurate measurements over the UV/VIS range. The light source consists of a Xenon flash lamp for the ultraviolet (UV) as well as for the visible (VIS) and near-infrared wavelength regions covering a spectral range from 190 up to 1100 nm. The lamp flashes are focused on a glass fiber which drives the beam of light onto a cuvette containing the sample solution. The beam passes through the sample and specific wavelengths are absorbed by the sample components. The remaining light is collected after the cuvette by a glass fiber and driven into a spectrograph. The spectrograph consists of a diffraction grating that separates the light into the different wavelengths, and a CCD sensor to record the data, respectively. The whole spectrum is thus simultaneously measured, allowing for fast recording.[16]
Samples for UV/Vis spectrophotometry are most often liquids, although the absorbance of gases and even of solids can also be measured. Samples are typically placed in a transparent cell, known as a cuvette. Cuvettes are typically rectangular in shape, commonly with an internal width of 1 cm. (This width becomes the path length, , in the Beer–Lambert law.) Test tubes can also be used as cuvettes in some instruments. The type of sample container used must allow radiation to pass over the spectral region of interest. The most widely applicable cuvettes are made of high quality fused silica or quartz glass because these are transparent throughout the UV, visible and near infrared regions. Glass and plastic cuvettes are also common, although glass and most plastics absorb in the UV, which limits their usefulness to visible wavelengths.[2]
Specialized instruments have also been made. These include attaching spectrophotometers to telescopes to measure the spectra of astronomical features. UV–visible microspectrophotometers consist of a UV–visible microscope integrated with a UV–visible spectrophotometer.
A complete spectrum of the absorption at all wavelengths of interest can often be produced directly by a more sophisticated spectrophotometer. In simpler instruments the absorption is determined one wavelength at a time and then compiled into a spectrum by the operator. By removing the concentration dependence, the extinction coefficient (ε) can be determined as a function of wavelength.
Microspectrophotometry[edit]
UV–visible spectroscopy of microscopic samples is done by integrating an optical microscope with UV–visible optics, white light sources, a monochromator, and a sensitive detector such as a charge-coupled device (CCD) or photomultiplier tube (PMT). As only a single optical path is available, these are single beam instruments. Modern instruments are capable of measuring UV–visible spectra in both reflectance and transmission of micron-scale sampling areas. The advantages of using such instruments is that they are able to measure microscopic samples but are also able to measure the spectra of larger samples with high spatial resolution. As such, they are used in the forensic laboratory to analyze the dyes and pigments in individual textile fibers,[17] microscopic paint chips[18] and the color of glass fragments. They are also used in materials science and biological research and for determining the energy content of coal and petroleum source rock by measuring the vitrinite reflectance. Microspectrophotometers are used in the semiconductor and micro-optics industries for monitoring the thickness of thin films after they have been deposited. In the semiconductor industry, they are used because the critical dimensions of circuitry is microscopic. A typical test of a semiconductor wafer would entail the acquisition of spectra from many points on a patterned or unpatterned wafer. The thickness of the deposited films may be calculated from the interference pattern of the spectra. In addition, ultraviolet–visible spectrophotometry can be used to determine the thickness, along with the refractive index and extinction coefficient of thin films.[8] A map of the film thickness across the entire wafer can then be generated and used for quality control purposes.[19]
Additional applications[edit]
UV/Vis can be applied to characterize the rate of a chemical reaction. Illustrative is the conversion of the yellow-orange and blue isomers of mercury dithizonate. This method of analysis relies on the fact that concentration is linearly proportional to concentration. In the same approach allows determination of equilibria between chromophores.[20][21]
From the spectrum of burning gases, it is possible to determine a chemical composition of a fuel, temperature of gases, and air-fuel ratio.[22]
See also[edit]
- Isosbestic point important in kinetics measurements as a control. A wavelength where absorption does not change as the reaction proceeds.
- Ultraviolet–visible spectroscopy of stereoisomers
- Infrared spectroscopy and Raman spectroscopy are other common spectroscopic techniques, usually used to obtain information about the structure of compounds or to identify compounds. Both are forms of vibrational spectroscopy.
- Fourier-transform spectroscopy
- Near-infrared spectroscopy
- Vibrational spectroscopy
- Rotational spectroscopy
- Applied spectroscopy
- Slope spectroscopy
- Benesi–Hildebrand method
- DU spectrophotometer – first UV–Vis instrument
- Charge modulation spectroscopy
References[edit]
- ^ a b Carroll, Gregory T.; Dowling, Reed C.; Kirschman, David L.; Masthay, Mark B.; Mammana, Angela (2023). «Intrinsic fluorescence of UV-irradiated DNA». Journal of Photochemistry and Photobiology A: Chemistry. 437: 114484. doi:10.1016/j.jphotochem.2022.114484.
- ^ a b c Skoog, Douglas A.; Holler, F. James; Crouch, Stanley R. (2007). Principles of Instrumental Analysis (6th ed.). Belmont, CA: Thomson Brooks/Cole. pp. 169–173. ISBN 978-0-495-01201-6.
- ^ a b R. S. Drago (1992). Physical Methods for Chemists, 2nd Edition. W. B. Saunders. ISBN 0030751764.
- ^ Metha, Akul (13 December 2011). «Principle». PharmaXChange.info.
- ^ Metha, Akul (22 April 2012). «Derivation of Beer–Lambert Law». PharmaXChange.info.
- ^ Misra, Prabhakar; Dubinskii, Mark, eds. (2002). Ultraviolet Spectroscopy and UV Lasers. New York: Marcel Dekker. ISBN 978-0-8247-0668-5.[page needed]
- ^ Historically, the term «Optical Density» (OD) was used instead of AU. But it is also worth noting that what is usually measured is percent transmission (%T), a linear ratio, which is converted to the logarithm by the instrument for presentation.
- ^ a b Löper, Philipp; Stuckelberger, Michael; Niesen, Bjoern; Werner, Jérémie; Filipič, Miha; Moon, Soo-Jin; Yum, Jun-Ho; Topič, Marko; De Wolf, Stefaan; Ballif, Christophe (2015). «Complex Refractive Index Spectra of CH3NH3PbI3 Perovskite Thin Films Determined by Spectroscopic Ellipsometry and Spectrophotometry». The Journal of Physical Chemistry Letters. 6 (1): 66–71. doi:10.1021/jz502471h. PMID 26263093. Retrieved 16 November 2021.
- ^ a b c d Metha, Akul (14 May 2012). «Limitations and Deviations of Beer–Lambert Law». PharmaXChange.info.
- ^ «Stray Light and Performance Verification».
- ^ «Wavelength Accuracy in UV/VIS Spectrophotometry».
- ^ Berberan-Santos, M. N. (September 1990). «Beer’s law revisited». Journal of Chemical Education. 67 (9): 757. Bibcode:1990JChEd..67..757B. doi:10.1021/ed067p757.
- ^ Wittung, Pernilla; Kajanus, Johan; Kubista, Mikael; Malmström, Bo G. (19 September 1994). «Absorption flattening in the optical spectra of liposome-entrapped substances». FEBS Letters. 352 (1): 37–40. doi:10.1016/0014-5793(94)00912-0. PMID 7925937. S2CID 11419856.
- ^ Ansell, S; Tromp, R H; Neilson, G W (20 February 1995). «The solute and aquaion structure in a concentrated aqueous solution of copper(II) chloride». Journal of Physics: Condensed Matter. 7 (8): 1513–1524. Bibcode:1995JPCM….7.1513A. doi:10.1088/0953-8984/7/8/002. S2CID 250898349.
- ^ Sooväli, L.; Rõõm, E.-I.; Kütt, A.; et al. (2006). «Uncertainty sources in UV–Vis spectrophotometric measurement». Accreditation and Quality Assurance. 11 (5): 246–255. doi:10.1007/s00769-006-0124-x. S2CID 94520012.
- ^ reserved, Mettler-Toledo International Inc. all rights. «Spectrophotometry Applications and Fundamentals». www.mt.com. Retrieved 10 July 2018.
- ^ Forensic Fiber Examination Guidelines, Scientific Working Group-Materials, 1999, http://www.swgmat.org/fiber.htm
- ^ Standard Guide for Microspectrophotometry and Color Measurement in Forensic Paint Analysis, Scientific Working Group-Materials, 1999, http://www.swgmat.org/paint.htm
- ^ Horie, M.; Fujiwara, N.; Kokubo, M.; Kondo, N. (1994). «Spectroscopic thin film thickness measurement system for semiconductor industries». Conference Proceedings. 10th Anniversary. IMTC/94. Advanced Technologies in I & M. 1994 IEEE Instrumentation and Measurement Technology Conference (Cat. No.94CH3424-9). pp. 677–682. doi:10.1109/IMTC.1994.352008. ISBN 0-7803-1880-3. S2CID 110637259.
- ^ Sertova, N.; Petkov, I.; Nunzi, J.-M. (June 2000). «Photochromism of mercury(II) dithizonate in solution». Journal of Photochemistry and Photobiology A: Chemistry. 134 (3): 163–168. doi:10.1016/s1010-6030(00)00267-7.
- ^ UC Davis (2 October 2013). «The Rate Law». ChemWiki. Retrieved 11 November 2014.
- ^ Mekhrengin, M.V.; Meshkovskii, I.K.; Tashkinov, V.A.; Guryev, V.I.; Sukhinets, A.V.; Smirnov, D.S. (June 2019). «Multispectral pyrometer for high temperature measurements inside combustion chamber of gas turbine engines». Measurement. 139: 355–360. Bibcode:2019Meas..139..355M. doi:10.1016/j.measurement.2019.02.084. S2CID 116260472.