Расчёты в гравиметрическом анализе Расчёт массы навески исследуемого вещества
При расчётах массы
навески исходят из того, что масса
полученной весовой формы при взвешивании
на аналитических весах не должна
превышать относительную ошибку метода
![]() .
.
Исходя из опыта, установлены следующие
пригодные для работы массы навесок:
= для кристаллических
осадков – масса весовой формы не менее
0,5 г:
![]() ;
;
= для аморфных
осадков масса весовой формы – не менее
0,1 г:
![]() ,
,
где mн– масса навески анализируемого вещества,
г;![]() – приблизительное содержание исследуемого
– приблизительное содержание исследуемого
вещества;F–
гравиметрический (аналитический) фактор:
![]() ,
,
где Мв-ва– молярная масса определяемого элемента;Мв.ф.– молярная масса весовой
формы;z– число молей
анализируемого элемента в 1 моль
весовой формы. Например, для оксида
железа (III)Fe2O3коэффициентzдля
железа равен 2.
Расчет объёма раствора осадителя
При расчете объёма
раствора осадителя пользуются формулой:
![]() ,
,
где Voc— объём осадителя, мл; 1,5 – эмпирический
коэффициент избытка осадителя относительно
теоретически рассчитанного по уравнению
реакции осаждения; ω – концентрация
раствора осадителя, %;d– плотность раствора осадителя, г/см3;F– гравиметрический
(аналитический) фактор;mн— масса навески анализируемого вещества, г.
Расчеты результатов весового анализа при использовании метода осаждения
Вычисление массовой
доли элемента ведут по уравнению:
![]() ,
,
![]() –массовая доля
–массовая доля
анализируемого вещества, %; mв.ф.– масса весовой формы, г;F– гравиметрический (аналитический)
фактор;mн–
масса навески анализируемого вещества, г.
При расчёте
концентрации анализируемого элемента
в растворе, выраженной в г/л, используют
формулу:
![]() ,
,
где mв.ф.– масса весовой формы, г;F– гравиметрический (аналитический)
фактор;Vа– объем аликвоты, мл.
Объёмный (титриметрический) анализ
Данная группа
методов анализа основана на измерении
объёма раствора с известной концентрацией,
который необходимо добавить к пробе
известного объёма (аликвоте) для
протекания аналитической реакции в
соответствии со стехиометрическим
уравнением. Добавляемый реагент называют
титрантом, а его раствор с заданной
концентрацией – титрованным раствором.
В процессе титрования
к раствору исследуемого вещества
небольшими порциями прибавляют раствор
титранта. По мере его добавления
концентрация исследуемого раствора
уменьшается, а продуктов реакции –
увеличивается.
Момент титрования,
когда титрант добавлен в количестве,
эквивалентном исследуемому веществу,
называется точкой эквивалентности.
В химических
методах анализа точку эквивалентности
определяют визуально по аналитическим
эффектам: изменению цвета раствора,
образованию осадка и др. Для этой цели
используют специальные вещества –
индикаторы, которые способны менять
свой цвет в точке эквивалентности.
В физико-химических
методах анализа точку эквивалентности
устанавливают по резким изменениям
электропроводности, рН раствора и др.
Соответствующий
точке эквивалентности объём титранта
называют эквивалентным.
Относительная
ошибка объёмного анализа составляет
0,2 %.
Требования к
химическим реакциям объемного анализа:
= реакция должна
протекать быстро и быть практически
необратимой;
= взаимодействие
между определяемым веществом и титрантом
должно протекать стехиометрически,
т.е. в точном соответствии с уравнением
реакции;
= возможность
четкой фиксации точки эквивалентности;
= в растворе должны
отсутствовать вещества, мешающие ходу
основной реакции и определению конечной
точки титрования.
Преимущества
объемных методов анализа: скорость,
простота, точность и возможность
автоматизации.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Метрологические свойства весов
Весы, независимо
от их конструкции, должны иметь такие
метрологические свойства.
Устойчивость
— способность
весов, выведенных из состояния равновесия,
быстро возвращаться к первоначальному
положению.
Устойчивость весов
достигается при условии, если их коромысло
находится в состоянии устойчивого
равновесия. Устойчивость весов зависит
также от отдаленности центра от точки
опоры, причем, чем ниже размещен центр
весов, тем больше устойчивость и тем
труднее их вывести из состояния
равновесия, а соответственно ниже их
чувствительность.
Проверка устойчивости
проводится следующим образом: весы,
находящиеся в равновесии, необходимо
вывести из положения равновесия, слегка
коснувшись чашки. Подсчитать количество
колебаний стрелки весов до возвращения
ее в положение равновесия. Если стрелка
совершила не более 4—6 колебаний, можно
считать, что весы устойчивы.
Постоянство
показаний
— способность
весов показывать одни и те же результаты
при многоразовых определениях массы
тела, проводимых на этих весах в одних
и тех же условиях.
Проверка постоянства
показаний проводится следующим образом:
весы должны находиться в положении
равновесия. На технических аптечных
весах уравновешивают, например, флакон
с массой гирь. Если все 3 раза масса
взвешиваемого тела одна и та же, то весы
обладают постоянством показаний. Если
результаты не совпадают, то это значит,
что острия призм не вполне параллельны,
смещены. Кроме того, на постоянство
показаний большое влияние оказывает
величина трения в подвижных контактах
весов. Затупленность призм и их
загрязненность приводит к нарушению
постоянства показаний весов. Поэтому
весы в нерабочем состоянии должны
храниться так, чтобы избежать затупленности
призм.
Причиной непостоянства
показаний весов является несовершенство
устройства весов (например, незаметное
смещение отдельных частей при пользовании
ими), а также условия, в которых производится
взвешивание (например, одностороннее
нагревание коромысла (электрическая
лампа, солнечные лучи и пр.), вследствие
чего может произойти удлинение одного
плеча).
Чувствительность
— способность
весов показывать минимальное изменение
нагрузки в момент равновесия.
При этом чем меньше
разница в весе гирь, которую можно
определить этими весами, тем выше их
чувствительность и тем точнее результаты
взвешивания.
Чувствительность
весов прямо пропорциональна длине плеча
коромысла и обратно пропорциональна
массе коромысла, нагрузке весов (массе
чашек, груза, перегруза), величине прогиба
коромысла, расстоянию от точки опоры
до центра тяжести коромысла. Чувствительность
весов определяется формулой:
![]()
где
S
—
чувствительность, мм/мг;
L
— длина
плеча коромысла, мм;
Р —
масса чашки с грузом, мг;
р —
масса перегруза, выводящего весы из
состояния равновесия, мг;
h
— расстояние
от точки опоры до линии, соединяющей
острия грузоподъемных
и опорной
призм (величина прогиба коромысла), мм;
R
— масса
коромысла, мг;
т. —
расстояние от точки опоры до центра
тяжести коромысла, мм; Z
— длина
стрелки, мм;
X
— цена деления шкалы, мм.
Из формулы следует,
что величины L,
R,
m,
Z,
X
зависят от
конструкции весов.
В наибольшей
степени на величину чувствительности
весов влияет величина прогиба коромысла
h.
Весы с
прямолинейным коромыслом характеризуются
тем, что острия опорной и грузоприемных
призм находятся на одной прямой линии
(прямолинейность весов). При этом величина
h
= 0, а уравнение
принимает вид:
![]()
то есть чувствительность
весов становится независимой от величины
нагрузки весов. Это возможно лишь при
взвешивании минимальных количеств. На
практике даже незначительное определение
массы вызывает прогиб коромысла и влияет
на чувствительность весов. С увеличением
нагрузки весов, как следует из формулы,
их чувствительность уменьшается. Прогиб
коромысла при этом может быть недопустимо
большим, а коромысло необратимо
деформироваться, то есть весы выйдут
из рабочего состояния. Во избежание
этого необходимо во время взвешивания
не превышать величины предельно
допустимой нагрузки, обозначенной на
коромысле весов.
Чувствительность
весов зависит от ряда факторов.
От расположения
центра тяжести коромысла по отношению
к точке его опоры. Весы
будут иметь наибольшую чувствительность
при достаточной устойчивости в том
случае, когда центр тяжести коромысла
лежит как можно ближе к точке опоры, но
во всяком случае ниже этой точки.
От массы коромысла.
Чем меньше
масса коромысла, тем большую чувствительность
при всех прочих условиях будут иметь
весы. Для облегчения коромысла в нем
делаются вырезы круглой или иной формы.
От длины плеч
коромысла. Теоретически,
чем больше длина плеч коромысла, тем
больше его чувствительность. Однако
практически нецелесообразно изготовлять
коромысла с длинными плечами, так как
удлинение коромысла ведет к увеличению
его массы, что вызывает уменьшение
чувствительности. Наряду с увеличением
длины коромысла значительно возрастает
прогиб его от действия груза, вследствие
чего понижается центр тяжести коромысла
и чувствительность весов уменьшается.
Поэтому практически коромысла делают
с как можно более короткими плечами.
На чувствительность
весов оказывает влияние величина
трения между призмами и
подушками: чем больше трение, тем меньше
чувствительность, чем острее призмы,
тем чувствительность больше. Поэтому
призмы делают острыми из закаленной
стали. Износ-затупление призм приводит
к увеличению трения, и, следовательно,
к уменьшению чувствительности весов.
Следует отметить,
что большинство весов, служащих для
взвешивания с
большей
точностью (в том числе тарирные и ручные),
имеют длинные стрелки. Это объясняется
тем, что увеличение длины стрелки весов
позволяет наблюдать и отсчитывать
незначительные угловые отклонения
коромысла, так как чем больше радиус
(длина стрелки), тем больше длина дуги,
соответствующей отклонению коромысла
на один и тот же угол.
Проверка
чувствительности весов
проводится
путем определения минимальной нагрузки
(мг), вызывающей стандартное отклонение
стрелки от положения равновесия. За
стандартное отклонение принимают
отклонение стрелки, равное 5 мм (или трем
делениям шкалы) для тарирных весов и
выход стрелки из обоймицы до половины
своей длины с образованием угла, равного
приблизительно 5°, для ручных весов.
Чувствительность,
выраженную абсолютным значением груза,
вызывающего стандартное отклонение
стрелки, называют абсолютной
чувствительностью (<Sa6c),
или абсолютной
ошибкой взвешивания.
Чувствительность
ручных и тарирных весов определяют в
трех положениях: нагруженных на 1/10
предельной нагрузки, предельно нагруженных
и ненагруженных. Если груз, соответствующий
величине допустимой погрешности
(установленной ГОСТом) для данного типа
весов (табл. 5), помещенный на одну из
чашек таких весов, вызывает стандартное
отклонение стрелки, то весы считаются
чувствительными .
Так весы технические
ВКТ-1000 имеют следующие величины
погрешностей:
— ненагруженных
— 20 мг;
— с 1/10 максимальной
нагрузки — 60 мг;
— максимально
нагруженные — 100 мг.
При определении
величины чувствительности этих весов
при 1/10 нагрузки поступают так: весы
приводят в состояние равновесия, на
каждую чашку помещают гири массой по
100,0 г, уравновешивают весы, затем
постепенно нагружают правую чашу весов
гирьками (от меньшей к большей). Груз,
вызывающий стандартное отклонение
стрелки, показывает абсолютную
чувствительность. Если величина
дополнительной нагрузки больше допустимой
погрешности (60 мг), например 80 мг, весы
считают нечувствительными. Их изымают
из пользования и направляют на проверку
до срока клеймения. Аналогично проводят
определение чувствительности при
ненагруженных и при предельной нагрузке
весов. Чувствительность весов можно
определить и таким образом: при трех
состояниях весов, приведенных в положение
равновесия, на одну из чашек кладут гирю
массой, соответствующей величине
допустимой погрешности.
Таблица 5
Метрологическая
характеристика ручных и тарирных весов
|
Типоразмеры |
Нагрузка, г |
Допустимая |
|||
|
Максимальная |
Минимальная |
Ненагруженных |
При 1/10 предельной |
При максимальной |
|
|
ВР-1 |
1 |
0,02 |
2 |
3 |
5 |
|
ВР-5 |
5 |
0,010 |
2 |
4 |
10 |
|
ВР-20 |
20 |
1,00 |
3 |
6 |
20 |
|
ВР-100 |
100 |
5,00 |
5 |
10 |
50 |
|
ВКТ-1000 |
1000 |
50,00 |
20 |
60 |
100 |
|
Т-2-1000 |
1000 |
50,00 |
20 |
50 |
200 |
Если во всех трех
случаях стрелка весов отклонилась на
расстояние 5 мм (или 3 деления по шкале),
то весы обладают достаточной
чувствительностью. Если стрелка
отклонилась на расстояние менее, чем 5
мм, то такие весы недостаточно
чувствительные и ими пользоваться
нельзя.
Что касается
определения чувствительности ручных
весов, то оно осуществляется аналогичным
путем по стандартному отклонению
стрелки. В этих случаях чувствительность
весов выражается числом миллиграммов,
вызывающих необходимое отклонение
стрелки.
На практике большое
значение имеет так называемая относительная
чувствительность, которая может указать
относительную ошибку взвешивания
(точность дозирования).
Относительная
чувствительность весов Sотн
может
быть выражена отношением минимального
груза Р, вызывающего заметное отклонение
стрелки от нулевого положения шкалы, к
нагрузке л,
лежащей на
одной чашке весов, потому что
чувствительность весов может немного
варьировать в зависимости от величины
нагрузки:
![]() или
или
![]()
Если, например,
весы нагрузить гирями по 100,0 г на каждую
чашечку и поместить дополнительный
груз, равный 0,05 г, который дает стандартное
отклонение стрелки, то относительная
чувствительность весов равна:
![]()
Значит, на этих
весах можно взвешивать груз, равный
100,0 г, с точностью 0,0005 его настоящей
массы, то есть относительная ошибка не
превышает 0,05 % (0,0005 • 100). Такие весы можно
считать достаточно чувствительными.
Определение
ошибки взвешивания.
На одних и
тех лее весах груз можно взвесить с
различной точностью. Наибольшая точность
может быть получена тогда, когда навеска
близка по значению к наибольшей допустимой
нагрузке весов. Ошибка взвешивания
возрастает, если переходят границу
предельной или минимальной нагрузки,
которая обозначена на коромысле данных
весов.
Чтобы сделать
вывод, насколько правильно выбраны весы
для определения массы вещества, надо
установить точность взвешивания или
относительную ошибку (в %).
Например, необходимо
взвесить массу 0,06 г. Какими весами при
этом следует воспользоваться? Для весов
ВР-100 величина 0,06 г близка к ненагруженным
весам, поэтому Sабс
по табл. 5
равна 0,005 г, а SOTH
— 8%:
![]()
Для весов ВР-1
величина массы 0,06 г близка к ненагруженным
весам, поэтому Sа6с
(табл. 5) — 0,002 г, а
S отв
— 3,6 %:
![]()
Таким образом, для
взвешивания массы 0,06 г нужно использовать
ручные весы ВР-1.
Расчет относительной
ошибки взвешивания можно осуществить
путем составления соответствующей
пропорции.
Например, рассчитать
относительную ошибку при взвешивании
0,1 г натрия хлорида на ВР-1. По табл. 5
находят ошибку, допустимую при нагрузке
0,1 г. Так как навеска наиболее близка по
значению к 1/10 предельной нагрузки, а не
к предельной нагрузке или к ненагруженным
весам, то допустимая погрешность Soth
равна 0,003
(3 мг). Составив пропорцию, находят ошибку
взвешивания (х),
которая
составляет ±3 %:
0,1 – 0,003
100 — х
![]()
При взвешивании
на этих же весах большего количества
натрия хлорида 0,9 г (навеска наиболее
близка по значению к предельной нагрузке
ВР-1) допустимая погрешность равна 0,005
(5 мг). Относительная ошибка в этом случае
составляет ±0,55 %:
0,9 – 0,005
100 — х
![]()
Точность или
правильность
— способность
весов показывать правильное соотношение
между массой взвешиваемого вещества
и соответствующими гирями.
Точность или
правильность весов зависит от таких
факторов:
— от равноплечности
коромысла весов;
—от параллельности
острия опорной и грузопринимающих
призм;
— от положения
центра тяжести весов коромысла, который
должен лежать точно на вертикали,
проходящей через точки опоры, которые
находятся ниже нее;
— от равенства
массы чашек.
Если весы отвечают
указанным требованиям, то их коромысло
должно находиться в горизонтальном
положении, а показчик равновесия
(стрелка) — строго вертикальном как
при пустых, так и при нагруженных
одинаковым грузом чашечках. Вследствие
невозможности обеспечить у весов
абсолютно точное равенство плеч и в
связи с трением, создающимся в опорных
деталях коромысла при его колебаниях,
весы всегда имеют ограниченную точность.
В связи с этим для всех весов установлены
максимально допустимые погрешности и
весы считаются правильными (верными),
если их погрешности не превышают
установленных значений.
Проверка
точности весов
проводится
при 1/10 максимальной нагрузки, при полной
нагрузке и ненагруженных весах.
Например, необходимо
определить точность технических весов
с максимальной нагрузкой 1 кг. Для этого
на левую чашку помещают гирю, равную
1/10 максимальной нагрузки, 100,0 г, а на
правую — тарирный стакан с дробью и
добиваются равновесия. Затем при
нерабочем положении арретира гирю и
груз меняют местами и переводят арретир
в рабочее положение. Стрелка весов при
перемене местами гири и груза должна
прийти в положение равновесия. Если
равновесие восстанавливается, то весы
верны (равноплечи). При отсутствии
равновесия на поднявшуюся чашку весов
добавляют груз-допуск (миллиграммовый
разновес), равный величине погрешности
(см. табл. 5). Это должно привести весы в
состояние равновесия или отклонить
стрелку в противоположную сторону не
более чем на 5 мм. Вес добавленных гирь
в этом случае будет являться величиной
неравноплечно-сти весов. Если этого не
произойдет, весы не обладают достаточной
точностью, они неравноплечи. Неравноплечие
весы применять нельзя, так как они не
будут давать точные показатели массы
взвешиваемого вещества.
Гири и разновес.
Разновес представляет собой набор
гирь. Гири
— это
меры определенно установленной массы
(веса), служащие для измерения массы
тела по весу.
Взвешивая тело,
мы сравниваем его массу с величиной,
принятой за единицу по Международной
метрологической системе мер. За единицу
массы принимается килограмм. В
повседневной аптечной практике основной
единицей измерения массы лекарственного
средства является грамм — тысячная
доля килограмма. Названия низших единиц
долей грамма образуются с помощью
латинских приставок «деци» (0,1), «санти»
(0,01), «милли» (0,001). В рецепте слово «грамм»
или его обозначение «г» опускается.
Всякое число в рецепте, обозначенное
десятичными знаками, целыми или дробными,
принимается за выражение количества
вещества в граммах, если нет других
обозначений.
В зависимости от
назначения различают такие гири:
— образцовые,
изготовляемые из золота, платины и
медных сплавов;
— аналитические,
изготовляемые из медных сплавов и стали
с тщательно отполированной поверхностью,
покрытой золотом, платиной, никелем
или хромом;
— технические
1, 2 и 3-го классов.
В аптечной практике
применяются технические гири 2-го класса
в виде специальных наборов (разновесов):
крупного (граммового), содержащего гири
от 1,0 до 500,0 г, и мелкого (миллиграммового),
содержащего гири от 0,01 до 0,500 г. Граммовый
разновес изготавливают из латуни или
углеродистой стали с никелевым или
хромовым покрытием для предохранения
от окисления. Поверхность гирь должна
быть гладкой, без трещин, царапин и т.
п. Гири имеют форму прямых цилиндров с
головками. Миллиграммовые гири
изготавливают из мельхиора или алюминия
в виде разной формы пластинок:
треугольников, квадратов, шестиугольников.
Для защиты от
внешних влияний и повреждений гири
хранят в специальных коробках с гнездами
(рис. 15).
Гири должны
содержаться в чистоте, для чего
периодически их очищают от пыли и жира,
промывая в мыльной теплой воде или в
органических растворителях (спирте,
бензине), после чего тщательно вытирают
насухо
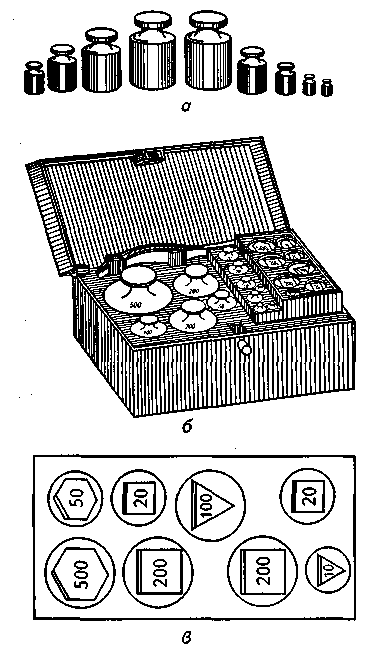
Рис. 15. Разновес:
а —
крупный; б
— набор в
футляре; в
— мелкий
мягкой тканью.
Брать их необходимо только пинцетом,
чистить гири
разными
полирующими средствами категорически
запрещается.
Гири и разновесы
подвергаются проверке и клеймению один
раз в год. Исправность весового хозяйства
аптечных учреждений контролируется
представителями местных отделений
Государственного комитета стандартов,
мер и измерительных приборов. При этом
на коромысло ручных и тарирных весов
наносится клеймо с указанием года
проверки (выбивают две последние цифры,
например, цифра 97 означает, что проверка
весов была проведена в 1997 г.).
Правила взвешивания
на технических и ручных равноплечих
весах. Прежде
чем начать взвешивание, нужно проверить
соответствие весов выше указанным
метрологическим свойствам, то есть
убедиться в их точности, чувствительности,
неизменности показаний и устойчивости.
Необходимо придерживаться предельной
и минимальной нагрузок, установленных
для данных весов.
Перед работой
весы следует осмотреть, протереть
марлевой салфеткой, смоченной
спирто-эфирной смесью, убедиться в их
равновесии в ненагруженном состоянии.
Если весы не уравновешены, то их
уравновешивают при помощи регуляторов,
помещенных на концах коромысла.
Как правило (для
удобства), гири помещают на левую чашечку
весов, а взвешиваемое лекарственное
средство — на правую.
При взвешивании
лекарственных и вспомогательных веществ
их названия ассистент читает трижды:
снимая с вертушки, при отвешивании и
возвращении штангласа на место. Подсчет
массы гирь производится дважды — в
начале взвешивания и по окончании.
Порошкообразные
вещества при взвешивании на ручных
весах помещают непосредственно на
чашку весов, а густые — на кружок
пергаментной или фильтровальной бумаги
(предварительно стари-рованный).
Взвешивать какие-либо вещества
непосредственно на чашечке тарирных
весов недопустимо, нужно применять
соответствующую тару (флаконы, банки,
капсулы и др.). Во избежание ошибок не
рекомендуется для тарирования применять
разновес.
Сыпучие вещества
отвешивают непосредственно из штангласа
путем легкого постукивания по нему
указательным пальцем правой руки.
Прибавляют вещество небольшими порциями
так, чтобы нити весов не загрязнялись.
По мере приближения момента равновесия
порции добавляемого вещества уменьшают,
чтобы исключить возможную передозировку
порошка. В случае необходимости вещество
отбирают при помощи пластмассовой или
целлулоидной пластинки. После взвешивания
с весов сначала снимают разновес (и
второй раз проводят подсчет массы
гирь), а затем лекарственное вещество.
После каждого отвешивания лекарственного
вещества из штангласа шейку и пробку
штангласа, а также чашечки весов
тщательно протирают марлевой салфеткой.
Пример. Определить относительную ошибку взвешивания 100 г хлористого кальция, если в результате взвешивания получено 99,8473 0,0001 г [c.222]
Вычислить относительную ошибку взвешивания осадка ( весовой формы ) при определении 1 г кальция в обоих случаях, если весы позволяют взвешивать с точностью 0,0001 г. [c.67]
Какую наименьшую навеску можно брать на аналитических весах с ценой деления шкалы для стрелки 0,3 мг при допустимой относительной ошибке взвешивания не более 0,3 /о (точку равновесия определяют с точностью 0,5 деления) [c.29]
Взвешивание — это одна из наиболее точных операций анализа. Относительная ошибка взвешивания зависит от взвешиваемой массы. Если взвешивание проводят с точностью до 0,1 мг и взвешивают не менее 100 мг (как это бывает в обычных анализах), то ошибка взвешивания ничтожно мала. [c.299]
Вычислим, с какой точностью определена эта концентрация, если взвешивание проводилось на аналитических весах с точностью до 0,0002 г, а измерение объема раствора содержит ошибку, равную Н-0,5 мл. Относительная ошибка взвешивания в данном случае равна [c.56]
Относительные ошибки взвешивания осадков будут в первом случае [c.176]
Недостатком соды в сравнении с бурой является меньший эквивалентный вес, в связи с чем увеличивается относительная ошибка взвешивания. Кроме того, безводная сода гигроскопична, и перед употреблением ее необходимо прокалить для удаления влаги. [c.330]
Например, при повторных взвешиваниях стеклянного фильтра были получены следующие результаты (в г) 10,2375 10,2374 10,2378 10,2375. Определить среднее арифметическое значение, дисперсию, среднюю квадратичную ошибку, среднюю квадратичную ошибку среднего арифметического, коэффициент нормирования отклонений, вероятное квадратичное отклонение среднего арифметического, истинную массу стеклянного фильтра и относительную ошибку взвешивания с надежностью а, равной 0,95. Для удобства вычислений запишем данные в виде таблицы [c.302]
Относительными ошибками взвешивания и измерений объемов по бюретке пренебрегаем следовательно, [c.256]
Более точные данные определений получаются с рабочим раствором буры, поскольку эквивалентный вес ее (190,72) в 3,6 раза больше, чем соды (53). При большом эквивалентном весе исходного вещества уменьшается относительная ошибка взвешивания. [c.242]
Определение очень малых количеств какого-либо иона выгоднее производить в виде соединения с возможно большим молекулярным весом еще и потому, что здесь будут меньше не только относительные ошибки взвешивания, но и другие ошибки. [c.42]
Из всех условий, обычно предъявляемых к исходным веществам, в микрохимическом анализе особенно желательна работа с веществом, обладающим более высоким грамм-эквивалентом. Чем выше грамм-эквивалент, тем точнее может быть взята его навеска на микровесах. Если необходимо произвести определение с ошибкой до 0,1%, то ошибка в приготовлении раствора исходного вещества, являющаяся частью допустимой ошибки, не должна превышать 0,03%. Если учесть, что на обычных микровесах можно взвешивать со средней ошибкой +0,003 мг, то нетрудно вычислить, что минимальная навеска исходного вещества, при которой относительная ошибка взвешивания не превысит 0,03%, равна 10 мг. На титрование этой навески должно уйти в среднем 4 Л1Л 0,01 н. раствора, т. е. 0,04 мг-экв. Отсюда нетрудно рассчитать, что минимальная величина 1 г-экв должна быть около 250 г [c.162]
В нашей лаборатории применялись ультрамикровесы с нихромовым коромыслом длиною 250 мм и толщиною 0,2 мм для отбора навесок от 20 у ДО 600 т с относительной ошибкой взвешивания не более 3—5%. [c.244]
Иметь достаточно высокую эквивалентную массу. Масса вещества, требующегося для стандартизации или приготовления раствора данной концентрации, тем больше, чем выше его эквивалентная масса. Поскольку прп увеличении массы относительная ошибка взвешивания уменьшается, высокая эквивалентная масса способствует уменьшению ошибок взвешивания. [c.172]
Это означает, что 1 мг 5102 в пробе соответствует более 40 мг осадка крем- немолибденовой соли, что обеспечивает относительную ошибку ( ) взвешивания 1 мг 5Юз не выше 0,5 %, если принять абсолютную погрешность взвешивания на аналитических весах примерно 0,2 мг. [c.26]
Сказанное справедливо, однако, только при условии, если не приходится брать для титрования слишком малых навесок, так как это сильно увеличивает относительную ошибку взвешивания. [c.224]
Относительная ошибка определения мольной доли искусственно введенной примеси не превосходит суммы относительной ошибки взвешивания исследуемого вещества и относительной ошибки взвешивания искусственной примеси. В нашем случае обычно величина навески вещества составляет 1—1,5 г, навеска примеси — 0,01 г, погрешность взвешивания 0,0001 г. Из этого следует, что такой относительной ошибкой взвешивания исследуемого вещества можно пренебречь, так как она будет примерно на два порядка меньше относительной ошибки взвешивания искусственной примеси. Ошибку определения мольной доли искусственной примеси будет определять величина относительной ошибки взвешивания искусственной примеси. Из этого вытекает, что при такой точности взвешивания не следует брать навеску примеси меньше 0,01 г, так как это приведет к значительной ошибке. [c.122]
Считая, что максимальная относительная ошибка взвешивания не должна превышать 0,25—0,337о, можно установить следующую зависимость между чувствительностью весов и минимальной навеской [c.23]
Относительная ошибка взвешивания обычно составляет г0,1°/о. т. е. Д у= =+гО,17о- [c.23]
Можно приблизительно вычислить желательную величину коэфициента пересчета для весового микрохимического анализа. Величина весовой формы определяется по разности двух взвешиваний (например, вес пустого тигля и вес тигля с весовой формой). Средняя ошибка каждого взвешивания на микровесах составляет 0,005 мг значит, ошибка определения веса весовой формы около 0,01мг. При навеске в 10 жг относительная ошибка взвешивания равна 0,1 /о, при 1 мг — достигает 1%. Если коэфициент пересчета равен 0,3, то ошибка весового определения достигает в этих случаях 0,03% и 0,3%, а при коэфициенте пересчета 0,1 ошибки соответственно равны 0,01 % и 0,1 %. Для 1—2мг весовой формы желательно, чтобы коэфициент пересчета не превышал 0,2. [c.42]
Отсюда можно заключить, что при прочих равных условиях определение кальция в виде СаС204-Н20 (где фактор пересчета меньший) точнее, так как относительная ошибка взвешивания в этом случае меньше. [c.236]
Фактор пересчета для определения кальция в виде СаО равен 0,7147, а для определения в виде СаС204 Н2О равен 0,2743. Вычислить относительную ошибку взвешивания осадка ( весовой формы ) при определении 1 2 кальция в обоих случаях, если весы позволяют взвешивать с точностью + 0,000 1 г. [c.64]
На практике широко используются два метода конечного определения масла в органическом растворителе 1) весовой, в основе которого лежит определение масла взвешиванием после испарения органического растворителя, 2) различные варианты спектрального метода, в частности люминесцентный, а также по ИК- и УФ-спектрам поглощения. Весовой метод методически прост и надежен. Однако ои оказывается неэффективным при малом содержании масла в техническом продукте, когда относительные ошибки взвешивания стано-., вятся больщими. Основные требования, которые предт.явля-ются к органическому растворителю в весовом методе,— это нерастворимость технического продукта в экстрагенте и по (ВОЗМОЖНОСТИ низкая температура его кипения. В случае мочевины наиболее полно обоим требованиям удовлетворяет четыреххлористый углерод. [c.29]
Гравиметрический (весовой) метод анализа
Классическое название метода — весовой анализ. Гравиметрический анализ широко используют при количественных определениях. С его помощью определяют, например, содержание фосфора в фосфоритах, апатитах, фосфорных удобрениях, почвах, кормах и т. п.
1. Общая характеристика метода
Гравиметрией называют метод количественного анализа, заключающийся в точном измерении массы определяемого компонента пробы, выделенного в виде соединения известного состава или в форме элемента.
Гравиметрический анализ основан на законе сохранения массы веществ при химических превращениях. Это наиболее точный из химических методов анализа, его характеристики: предел обнаружения — 0,10%; правильность — 0,2 отн.%; информативность — 17 бит. В гравиметрии используют реакции обмена, замещения, разложения и комплексообразования, а также электрохимические процессы. Наиболее распространен метод осаждения.
1. Метод осаждения – это метод, при котором навеску анализируемого вещества растворяют и прибавляют 1,5-кратный избыток реагента- осадителя, соблюдая необходимые условия осаждения. Полученный осадок называют осаждаемой формой. Осадок отделяют от раствора (чаще всего фильтрованием), промывают, затем высушивают или прокаливают, получая гравиметрическую (весовую) форму. Массу определяемого компонента mа рассчитывают по формуле:
ma =mBF •100/а (%)
где mа — масса высушенного или прокаленного осадка, г;
F — гравиметрический фактор, определяемый по химической формуле гравиметрической формы;
а — навеска анализируемого вещества, г.
Гравиметрические факторы, называемые также аналитическими множителями или факторами пересчета, вычисляют как отношение молекулярной массы определяемого компонента к молекулярной массе гравиметрической формы с учетом стехиометрических коэффициентов.
Пример. Вычислить гравиметрические факторы для следующих гравиметрических определений:
| Определяемый компонент | Al | Ca | CO2 | Ba |
| Гравиметрическая форма | Al2O3 | CaO | BaCO3 | BaSO4 |
Решение: F = ABa / MBaSO4= 137.4 / 233.4 = 0.5887
2. Методы выделения — основаны на выделении определяемого компонента из анализируемого вещества и точном взвешивании его. Например, при определении содержания золы в твердом топливе сжигают определенное количество (навеску) этого топлива, взвешивают золу и вычисляют процентное содержание ее во взятом образце.
3.Метод отгонки состоит в том, что определяемый компонент «количественно» выделяют в виде летучего соединения (газа, пара) действием кислоты, основания или высокой температуры на анализируемое вещество. Например, определяя, содержание двуокиси углерода в карбонатной породе, обрабатывают образец ее соляной кислотой. Выделившийся газ пропускают через поглотительные трубки со специальными реактивами. По увеличению массы поглотительной трубки определяют количество выделившегося CO2.
4.Термогравиметрия. Выполнение большинства операций гравиметрического анализа (фильтрование, высушивание и прокаливание осадка, доведение его до постоянной массы) отнимает очень много времени. Однако с помощью термовесов, сконструированных Дювалем, удается значительно ускорить определение. В этом приборе можно нагревать твердые вещества до температуры приблизительно 10000C и наблюдать, как изменяется их масса. При этом прибор автоматически вычерчивает на бумаге кривую изменения массы вещества. Получающаяся ступенчатая кривая характеризует изменение массы осадка в процессе повышения температуры и даже позволяет судить о химических превращениях веществ.
Например, такая кривая показывает, что кристаллогидрат оксалата кальция CaC2O4•H2O устойчив лишь при температуре не выше 1000C. При повышении температуры до 2260C он разрушается с образованием безводной соли CaC2O4. Последняя при 4200C разлагается с получением карбоната кальция СаСО3. Далее при 6600C начинается распад карбоната на окись кальция и двуокись углерода. Этот процесс заканчивается при температуре 8400C.
2.Основные операции весового анализа
В ходе гравиметрического определения различают следующие операции: 1) отбор средней пробы вещества и подготовку ее к анализу; 2) взятие навески; 3) растворение; 4) осаждение определяемого элемента (с пробой на полноту осаждения); 5) фильтрование; 6) промывание осадка (с пробой на полноту промывания); 7) высушивание и прокаливание осадка; взвешивание; 9) вычисление результатов анализа.
Отбор средней пробы. Аналитическое определение лишь тогда приводит к содержательным выводам, когда отобранная для анализа проба является представительной по отношению к исследуемому материалу.
В производстве бывает необходимо определить средний химический состав большой партии неоднородного материала (удобрения, ядохимиката, почвы, руды и т. п.). При этом подготовка вещества к анализу сводится к правильному отбору так называемой средней пробы. Правила отбора средних проб различных материалов предусмотрены государственными стандартами или техническими условиями. Выполнение этой операции всегда подчинено единому принципу: средняя проба должна быть составлена из большого числа мелких порций, взятых в разных местах анализируемого материала. Благодаря этому состав отобранной пробы приближается к среднему химическому составу большого количества исследуемого материала.
Первичная средняя проба, отобранная тем или иным способом, еще непригодна для анализа. Обычно она слишком велика (от одного до нескольких килограммов) и неоднородна. Подготовка пробы состоит в измельчении, перемешивании и сокращении до небольшой массы (около 300 г). Для сокращения пробы пользуются так называемым квартованием . Измельченный материал перемешивают в куче, рассыпают ровным слоем в виде квадрата (или круга), делят на четыре сектора, содержимое двух противоположных секторов отбрасывают, а двух остальных — соединяют вместе. Операцию квартования повторяют многократно. Из полученного таким образом однородного материала берут навески для анализа.
Перекристаллизация. В условиях исследовательской лаборатории часто требуется найти содержание какого-нибудь элемента в химически чистом соединении (например, содержание бария в хлориде барии ВаС12•2Н20). Здесь подготовка вещества к анализу состоит и очистке его от примесей и обычно осуществляется путем перекристаллизации для удаления примесей только из кристаллических веществ, например из солей.
Применительно к пробоотбору введены следующие количественные характеристики:
1. Рабочий диапазон. A=mi — диапазон количеств определяемого компонента i, к которым применима данная методика.
2. Диапазон количества пробы P=mi+mo — диапазон общих количеств пробы, состоящий из определяемого компонента (индекс i) и «матрицы» (индекс 0) — суммы остальных компонентов. В зависимости от требуемого для анализа количества пробы методики обычно классифицируют следующим образом:

3.Диапазон содержаний компонента

В зависимости от величины G компоненты пробы обычно называют следующим образом:
| G> 10% | 10%>G> 1% | G< 1% |
| главный компонент | сопутствующий компонент | следовый компонент |
Легко видеть, что между A, P и G существует соотношение:
P = A/G• 100%
Отсюда можно оценить минимальное (и максимальное) количество пробы, требуемое для проведения анализа по выбранной методике, если заданы величины рабочего диапазона и содержание определяемого компонента. На практике следует по возможности брать количество пробы, несколько превышающее рассчитанное.
Примеры.
Содержание определяемого вещества в пробе приблизительно 10%; методика позволяет определять не менее 0.5 мг этого вещества. Минимальное количество пробы, требуемое для анализа, составляет:
P = 0,5 мг/10•100=5мг
Максимальное количество пробы, которым располагает аналитик, составляет 10 мг, содержание определяемого компонента в ней около 0,2%. Следовательно, необходимо использовать методику, позволяющую определять не менее:
А = 0,2 • 10 мг/100 = 0,02 мг = 20 мкг
2. Взятие навески
Навеской называют количество вещества, необходимое для выполнения анализа.
Как правило, чем больше навеска, тем выше и относительная точность определения. Однако работа с большой навеской имеет свои отрицательные стороны: получающийся при этом большой осадок трудно отфильтровать, промыть или прокалить, анализ занимает много времени. Наоборот, при слишком малой навеске ошибки взвешиваний и других операций, неизбежные при анализе, значительно снижают точность определения.
Таким образом, выбор величины навески анализируемого вещества определяется количеством осадка, наиболее удобным в работе. Например, на бумажном фильтре диаметром 7 см можно легко отфильтровать 0,5 г кристаллического сульфата бария ВаSО4. Но с таким же количеством аморфных, студенистых осадков гидрооксидов Fe(OH)3, Al(OH)3 работать чрезвычайно трудно.
Аналитической практикой установлено, что наиболее удобны в работе кристаллические осадки с массой около 0,5 г и объемистые аморфные осадки с массой 0,1—0,3 г. Учитывая эти нормы осадков и зная приблизительное содержание определяемого элемента в веществе, вычисляют необходимую величину навески.
Пример. Какую навеску хлорида бария BaCl2• 2H2O нужно взять для определения содержания в нем бария?
Исходные данные: формула осадка BaSO4; норма кристаллического осадка 0,5 г.
Решение. Величину навески находят из пропорции:
| 233,43 г | BaSO4 | получаются из | 244,31 г | BaCl2•2H2O |
| 0,5 г | BaSO4 | получаются из | Х г | BaCl2•2H2O |
X= 0,52 г.
Ответ: для анализа следует взять навеску хлорида бария ВаС12•2Н2О около 0,5— 0,6 г.
Иногда, выбирая навеску, учитывают необходимую точность определения и возможные потери из-за растворимости осадка.
Разумеется, выбор навески зависит еще от метода, с помощью которого будет выполняться определение (макро-, полумикро- или микроанализ). Мы рассмотрели случай выбора навески при макроанализе.
При определениях, не связанных с получением осадка, например при изучении влажности или зольности различных материалов, допустимы навески в 1,0—2,0 г, а иногда и больше. Вещество взвешивают в специальном стаканчике — бюксе.
3. Растворение навески анализируемого вещества
Для растворения навеску анализируемого вещества переносят (т. е. осторожно пересыпают) в чистый химический стакан нужного объема. Подходящий растворитель подбирают заранее, делая пробы с отдельными порциями вещества. Если предварительной пробой было установлено, что анализируемое вещество растворимо в воде, то навеску растворяют в 100—150 мл дистиллированной воды. При необходимости содержимое стакана нагревают на асбестированной сетке или на водяной бане, накрыв стакан часовым стеклом и не допуская кипения раствора.
Если исследуемое вещество не растворимо в воде, то навеску переводят в раствор действием кислоты (уксусной, соляной, серной, азотной) или царской водки. Количество той или иной кислоты, необходимое для растворения, вычисляют (с учетом ее концентрации) по уравнению реакции.
Выбор кислоты для растворения навески определяется, кроме того, характером происходящей при этом реакции. Например, известняк СаСО3 следует растворять в соляной кислоте, а не в серной, так как при действии последней образуется малорастворимый сульфат кальция CaSO4.
Навеску растворяют в кислоте осторожно, накрыв стакан часовым стеклом, чтобы избежать потери анализируемого вещества. Потеря возможна потому, что выделяющиеся газы (CO2, H2, H2S) увлекают с собой капельки раствора.
Приготовленный тем или иным способом раствор нередко приходится еще подготовить к анализу: упарить, нейтрализовать избыток кислоты, связать или удалить ионы, мешающие определению.
Разложение малорастворимых неорганических веществ. Разложение пробы малорастворимого неорганического вещества осуществляют «мокрым» путем (действием кислот) или «сухим» путем (сплавлением с карбонатом натрия, едкими щелочами и другими плавнями).
Азотная кислота как сильный окислитель растворяет медь, серебро, ртуть, мышьяк, висмут, бор, кадмий, германий, ванадий, марганец, молибден и некоторые другие металлы.
В концентрированной серной кислоте растворяют сплавы олова, сурьмы, свинца, а также ферротитан.
«Нержавеющие» (легированные) стали растворяют в хлорной кислоте.
Золото и платину растворяют в царской водке (конц.НСl и конц. НNО3), которая действует и как окислитель, и как комплексообразователь; в результате такой обработки получаются комплексные хлориды этих металлов. Вольфрамовые сплавы, молибден и ферромолибден, цирконий и тантал растворяются в смеси азотной и плавиковой кислоте образованием комплексных фторидов.
Пробы кремнекислоты, горных пород и различных силикатов разлагают действием плавиковой кислоты.
Перечисленные способы разложения анализируемой пробы и получения раствора не универсальны, непригодны для всех случаев анализа. Наиболее подходящий метод выбирают исходя из особенностей анализируемого материала.
4. Осаждение
Осаждение считают важнейшей операцией гравиметрического анализа.
При выполнении ее необходимо правильно выбрать осадитель, рассчитать его количество, соблюсти определенные условия осаждения, убедиться в полноте осаждения иона из раствора.
Выбор осадителя. Осадитель выбирают, исходя из ряда требований, предъявляемых к осадку.
1.Получающийся осадок (так называемая осаждаемая форма) должен прежде всего обладать как можно меньшей растворимостью в воде. Например, ион Ba2+ образует несколько малорастворимых солей: карбонат, оксалат, хромат и сульфат. Учитывая произведения растворимости их:
BaC2O4 — 1,6•10-10; BaCO3 — 2,4 • 10-10; ВаСО3 — 8,0•10-10; BaSO4 — 10-10. Очевидно, что при гравиметрическом определении ионы Ва2+ следует осаждать в виде сульфата BaSO4, имеющего наименьшую величину произведения растворимости.
2. Кроме того, получаемый осадок должен легко отфильтровываться и хорошо отмываться от примесей. Эти свойства наиболее характерны для крупнокристаллических осадков.
3. Наконец, осаждаемая форма должна при прокаливании полностью превращаться в весовую форму. Состав весовой формы должен точно соответствовать определенной химической формуле иначе невозможно провести вычисление результатов анализа. Например, осадок гидроокиси железа Fe(OH)3 в результате прокаливания полностью переходит в оксид железа Fe2O3. Последнюю и называют весовой формой и именно ее взвешивают в конце анализа.
Помимо этого, весовая форма не должна изменять своей массы на воздухе из-за поглощения паров воды и двуокиси углерода или вследствие частичного разложения. Для точности определения желательно также, чтобы весовая форма имела возможно большую молекулярную массу и содержала как можно меньше атомов определяемого элемента в молекуле. При этом погрешности определения (ошибки взвешивания, потери при перенесении осадка на фильтр и т. п.) меньше сказываются на результате анализа.
4.Кроме всех этих требований, предъявляемых к осадку при выборе осадителя, учитывают летучесть последнего. В качестве осадителя всегда предпочитают более летучее вещество, если примеси его не будут полностью удалены из осадка промыванием, то они улетучатся при последующем прокаливании. Например, для осаждения Ba2+ в виде сульфата бария пользуются серной кислотой, а не ее растворимыми солями (К2S04), так как кислота более летуча. По тем же соображениям ион Fe3+ осаждают из раствора действием летучего NH4OH, а не NaOH или KOH.
5. Выбираемый осадитель должен в той или иной мере обладать селективностью по отношению к осаждаемому иону. В противном случае приходится предварительно удалять другие ионы, мешающие определению. Такая селективность особенно характерна для органических реагентов, находящих применение не только в качественном, но и в количественном анализе.
Расчет количества осадителя. Необходимое количество осадителя вычисляют исходя из содержания осаждаемого иона в растворе и величины навески анализируемого вещества.
Пример. Для количественного определения Ba2+ растворили навеску BaCl2•2H2O в 0,4526 г. Какой объем 2 н. раствора серной кислоты потребуется для полного осаждения ионов Ba2+?
Решение: Из уравнения реакции видно, что одна грамм-молекула хлорида бария взаимодействует с одной грамм-молекулой серной кислоты:
BaCl2•2H2O + H2SO4 = BaSO4 + 2HC1 + 2H2O
Следовательно, на взаимодействие с 244,31 г BaCl2•2H2O расходуется 98 г H2SO4, которые содержатся в 1000 мл 2 н. раствора кислоты. Поэтому можно составить следующую пропорцию:
| на 244,31 г | BaCl2•2H2O | идет | 1000 мл | 2 н. H2SO4 |
| 0,4526 г | BaCl2•2H2O | идет | x мл | 2 н. H2SO4 |
Отсюда
х = (0,4526 • 1000)/244,31 =2 мл
Казалось бы, в этом примере для полного осаждения ионов Ba2+ достаточно взять 2 мл 2 н. H2SO4. Однако это не так. Абсолютно нерастворимых веществ не существует, и над осадком сульфата бария в растворе еще будут находиться неосажденные ионы Ba+. Поэтому необходимо принять меры, чтобы понизить концентрацию их в растворе, т. е. добиться практической полноты осаждения ионов Ba2+.
Известно, что жидкость над осадком представляет собой насыщенный раствор электролита. Произведение концентраций его ионов при неизменной температуре сохраняет постоянное значение, равное произведению растворимости. В рассматриваемом примере
ПРBaSO4 = [Ва2+] [SO42- ] = 1,1•10-10
Следовательно, чтобы понизить концентрацию ионов Ba2+, еще остающихся в растворе после осаждения, нужно повысить концентрацию других ионов (SO42-), т. е. действовать избытком осадителя (серной кислоты).
Опытным путем установлено, что для практически полного осаждения иона достаточно полуторного избытка осадителя. Добавление большего избытка осадителя может повысить растворимость осадка вследствие образования комплексных соединений, кислых солей и т. д.
Ответ: в рассматриваемом случае для полного осаждения ионов нужно взять не 2 мл, а 3 мл 2 н. H2SO4.
Наиболее благоприятные условия получения кристаллических и аморфных осадков неодинаковы.
При осаждении в растворе протекают два взаимосвязанных процесса: возникновение мельчайших зародышевых кристаллов и их дальнейший рост. Следовательно, надо по возможности уменьшить число центров кристаллизации и усилить рост уже образовавшихся кристаллов. Для достижения этих целей необходимо, чтобы раствор был возможно менее пересыщенным по отношению к осаждаемому соединению. Действительно, из сильно пересыщенного раствора осаждается множество мельчайших зародышевых кристаллов, которые почти не укрупняются. И, наоборот, в мало пересыщенном растворе создаются условия для дальнейшего роста небольшого количества образовавшихся кристаллов.
Возможно малое пресыщение раствора и получение крупнокристаллического осадка достигается при соблюдении особых условий. Разумеется, даже при соблюдении этих условий, помимо крупных кристаллов, получается и некоторое количество мелких. Чтобы их было меньше, осадок оставляют стоять на несколько часов (или до следующего занятия) для созревания (старения). Известно, что мелкие кристаллы любого вещества растворяются несколько быстрее, чем крупные, так как имеют большую поверхность соприкосновения с растворителем. Поэтому при созревании мелкие кристаллы растворяются и за их счет растут крупные. Поскольку крупные кристаллы имеют меньшую поверхность, соосаждение примесей понижается. Более быстрому созреванию осадка содействует повышение температуры, ускоряющее движение ионов в растворе. Поэтому стакан с осадком обычно оставляют в теплом месте, например на горячей водяной бане.
Проба на полноту осаждения. Как только раствор над осадком становится совершенно прозрачным, делают пробу на полноту осаждения иона. Для этого по стенке стакана прибавляют еще 2—3 капли раствора осадителя. Если при этом в месте смешения растворов появится хотя бы легкая муть, то считают, что полнота осаждения не достигнута. В таком случае добавляют к жидкости еще несколько миллилитров осадителя, перемешивают стеклянной палочкой, снова нагревают и оставляют стоять для созревания осадка. Иногда пробу на полноту осаждения приходится повторить несколько раз. Ее рекомендуется сделать и перед самым фильтрованием.
5. Фильтрование
Фильтрованием отделяют полученный осадок от раствора, содержащего посторонние примеси. Тщательность выполнения этой операции сказывается на точности определений.
В гравиметрическом анализе применяют не обычную фильтровальную бумагу, а так называемые беззольные фильтры. В процессе изготовления их промывают кислотами (HCl), удаляя большую часть минеральных веществ. Масса золы, образующейся при сжигании одного беззольного фильтра, бывает мала, поэтому ею пренебрегают.
Промышленность выпускает беззольные фильтры нескольких сортов, различающиеся по диаметру и плотности.
Черная (или красная) лента – наименее плотные, т.е. быстрофильтрующие и крупнопористые и используют для отделения аморфных осадков гидроксидов железа, алюминия и др.
Белая лента — фильтры средней плотности, применяемые для отделения большинства кристаллических осадков
Синяя лента — фильтры мелкопористые, наиболее плотные и медленно фильтрующие; применяют их для отделения мелкокристаллических осадков сульфата бария BaSО4 , эти фильтры называют также «баритовыми».
Иногда для фильтрования используют фарфоровую воронку Бюхнера, на дно которой помещают бумажный фильтр. Через нее фильтруют также при помощи вакуум-насоса.
6.Соосаждение. Промывание осадка
Осадок увлекает с собой посторонние вещества из раствора. Это явление, называемое соосаждением, — одно из серьезных помех при выполнении гравиметрического определения. Можно выделить четыре основных вида соосаждений.
Окклюзия — процесс захвата примесей микрокомпонента внутрь растущих кристаллов осадка основного компонента. Удаление окклюдированных примесей из осадка представляет трудную задачу.
Изоморфное соосаждение — процесс образования «смешанных кристаллов» с ионами основного компонента и микрокомпонента, имеющими близкие радиусы. Например, осадок сульфата бария может увлекать с собой из раствора примеси перманганата калия, так как эти вещества изоморфны, т.е. образуют совместную пространственную кристаллическую решетку.
Соосаждение с образованием химических соединений с осаждаемым веществом и присутствующими в растворе примесями также довольно распространено. Если осаждать из раствора ионы Ba2+ действием серной кислоты, то вместе с ними соосаждаются и примеси Fe3+ в виде комплексного сульфата Ва3[Fe(SO4)3]. В таких случаях необходимо предварительное удаление примесей из раствора. Так, перед осаждением ионов Ba2+ примеси Fe3+ приходится осадить аммиаком и отфильтровать гидроокись железа.
Иногда для удаления примесей используют так называемое переосаждение. Например, осадок оксалата кальция CaC2O4, содержащий примеси оксалата магния MgC2O4, растворяют в соляной кислоте, нейтрализуют раствор и переосаждают ион Ca2+, т. е. повторяют осаждение его оксалатом аммония. Поскольку переосаждение происходит при значительно меньшей концентрации ионов Mg2+, чем в первый раз, осадок CaC2O4 оказывается практически свободным от примесей MgC3O4.
Соосаждение в результате поверхностной адсорбции примесей осадком особенно часто встречается при осаждении аморфных веществ, имеющих разветвленную поверхность (гидроксиды железа и алюминия, кремневая кислота и т. п.).
Но адсорбция — это обратимый процесс. При длительном промывании осадка той или иной жидкостью поглощенные им примеси могут быть десорбированы, вымыты и удалены. Десорбции содействует также применение горячей промывной жидкости. Задача промывания и состоит в удалении посторонних примесей, адсорбированных осадком из раствора.
Иногда осаждаемое вещество увлекает примеси из раствора в результате сочетания нескольких видов соосаждения (адсорбционная окклюзия, химическая окклюзия и т. п.).
При промывании необходимо исключить потери осажденного вещества. Поэтому выбор промывной жидкости определяется свойствами промываемого осадка.
Промывание разбавленным раствором осадителя. При промывании большинства осадков дистиллированной водой возможно частичное растворение их, приводящее к потере осажденного вещества. Во избежание потерь такие осадки промывают разбавленным раствором осадителя, т. е. в промывную жидкость вводят осаждающий ион. Например, осадок оксалата кальция CaC3O4, заметно растворимый в воде, промывают разбавленным раствором осадителя, т. е, оксалата аммония (NH4) C2O4.
Промывание раствором электролита-коагулятора. Если осажденное вещество склонно к пептизации, то возможна потеря его в результате прохождения коллоида через фильтр. Чтобы этого избежать, такой осадок промывают разбавленным раствором электролита-коагулятора, препятствующего пептизации. Электролитами-коагуляторами обычно служат летучие вещества, легко удаляющиеся при последующем прокаливании осадка. Так, аморфные осадки гидроксидов Fe(OH)3 и Al(OH)3 промывают разбавленным раствором нитрата аммония.
Промывание дистиллированной водой. Промывание водой возможно только в тех немногих случаях, когда промываемый осадок практически не растворяется в воде, не пептизируется и не гидролизуется. Так, например, осадок сульфата бария BaSO4 промывают на фильтре дистиллированной водой.
Когда повышение температуры не увеличивает потери осажденного вещества используют не холодную, а горячую промывную жидкость, так как нагревание ускоряет десорбцию примесей.
Самое промывание производят сначала декантацией, т. е. приливают в стакан с осадком 15—20 мл промывной жидкости, тщательно перемешивают, дают осадку осесть и сливают жидкость по палочке на фильтр. При таком способе отмывание примесей значительно ускоряется. Промывание декантацией обычно производят 3—4 раза. Затем осадок количественно, без потерь, переносят на фильтр. Для этого наливают в стакан небольшую порцию промывной жидкости, взмучивают осадок и полученную суспензию осторожно сливают на фильтр по стеклянной палочке. Выполняя эту операцию нельзя терять ни одной капли жидкости. Пользуясь промывалкой, многократно обмывают стенки стакана небольшими порциями промывной жидкости и каждый раз сливают ее на фильтр. Частицы осадка, приставшие к стенкам стакана, сначала тщательно оттирают резиновым наконечником палочки, смывают на фильтр, а затем следы осадка снимают кусочком фильтра, который помещают в ту же воронку. Стеклянную палочку также обтирают кусочком фильтра и помещают его в воронку с осадком.
Наконец, когда ни в стакане, ни на палочке больше не останется частиц осажденного вещества, приступают к промыванию осадка на фильтре. Промывают его большим числом маленьких порций жидкости, которой всякий раз дают полностью стекать. Это обеспечивает более быстрое удаление примесей, чем в случае больших порций жидкости. Попутно осадок смывают в нижнюю часть фильтра.
Повторив промывание 4—5 раз, делают пробу на полноту удаления примесей. Для этого собирают из воронки в пробирку небольшую порцию фильтрата и прибавляют к нему реактив, дающий характерную реакцию с удаляемым из осадка ионом. Например, выполняя пробу на полноту удаления Cl— из осадка BaSO4, берут 1—2 мл фильтрата, подкисляют его азотной кислотой и действуют нитратом серебра. Если муть хлорида серебра при этом не появляется, то промывание прекращают. Фильтрат при гравиметрических определениях обычно не анализируют и отбрасывают, если он совершенно прозрачен, т. е. не содержит частиц осадка.
Фильтрование и промывание осадка следует выполнять на одном и том же занятии; отфильтрованный осадок сильно высыхает при хранении и не поддается промыванию.
7. Высушивание и прокаливание осадка
Отфильтрованный и промытый осадок еще содержит влагу; обычно его высушивают и прокаливают. Эти операции позволяют получить вещество со строго определенным химическим составом.
Высушивание осадка. Осадок высушивают вместе с фильтром. Воронку с осадком накрывают листком влажной фильтровальной бумаги. Ее края плотно прижимают к наружной поверхности воронки, лишнюю бумагу удаляют. Получается бумажная крышечка, плотно сидящая на воронке и защищающая осадок от пыли.
После этого воронку с осадком следует поместить на 20—30 мин в сушильный шкаф, имеющий полки с круглыми отверстиями. В одно из них и вставляют воронку. Температуру в шкафу поддерживают не выше 90—105° С — при более сильном нагреве фильтр обугливается и распадается.
Прокаливают осадки в фарфоровых тиглях различных размеров. Прежде чем приступить к прокаливанию, необходимо узнать массу пустого тигля. Для этого тигель предварительно прокаливают до постоянной массы, т. е. до тех пор, пока масса его перестанет изменяться. Прокаливают тигли в электрической муфельной печи, в тигельной печи или на газовой горелке, но обязательно при тех же температурных условиях, при которых предполагается прокаливать осадок. О температуре прокаливания ориентировочно судят по цвету каления муфельной (тигельной) печи:
| Начало темно-красного каления …………………………… | ~525°С |
| Темно-красное каление…………………………………………. | –7000C |
| Светло-красное каление………………………………………… | –900 – 10000C |
| Светло-оранжевое каление …………………………………… | ~1200°С |
| Белое каление …………………………………………………….. | –13000C |
| Ослепительно-белое каление………………………………… | –1400 – 15000C |
Предназначенный для прокаливания тигель берут тигельными щипцами за край и помещают в муфельную печь. После 25—30 мин прокаливания его вынимают из печи, дают остыть на листе асбеста (или на гранитной плитке) и переносят в эксикатор. Последний закрывают крышкой не сразу, а спустя 1—2 мин; иначе при охлаждении в эксикаторе создается разрежение и крышку будет трудно открыть. Затем эксикатор относят в весовую комнату и оставляют на 15—20 мин, чтобы тигель принял температуру весов.
Взвесив тигель на аналитических весах, его снова прокаливают 15—20 мин, охлаждают в эксикаторе и повторяют взвешивание. Если результат последнего взвешивания будет отличаться от предыдущего не более чем на ±0,0002 г, считают, что тигель доведен для постоянной массы, т. е. подготовлен для прокаливания осадка. В противном случае тигель прокаливают, охлаждают и взвешивают еще раз. Результаты всех взвешиваний непременно записывают в лабораторный журнал.
Прокаливание осадка. Кристаллизационная, или конституционная вода, которую может содержать даже высушенный осадок, должна быть полностью удалена путем прокаливания. Кроме того, при прокаливании нередко происходит химическое разложение вещества. Например, оксалат кальция CaC2О4•Н2О, получаемый при осаждении ионов Са2+ оксалатом аммония, уже при высушивании теряет кристаллизационную воду:
CaC2O4 • H2O → CaC2O4 + H2O
При слабом прокаливании он выделяет окись углерода и превращается в карбонат кальция:
CaC2O4 → СО2 + СаСО3
Наконец, при сильном прокаливании карбонат кальция разлагается с образованием двуокиси углерода и окиси кальция:
CaCO3 → CaO + CO2
По массе окиси кальция и вычисляют результат определения. Температура и продолжительность прокаливания осадков могут быть различны.
В самой технике прокаливания различают два случая.
1. Прокаливание осадка без отделения фильтра. Этот способ используют, когда прокаливаемый осадок не взаимодействует с углеродом обуглившегося фильтра. Так, без удаления фильтра прокаливают осадки оксидов Al2O3, CaO и некоторые другие.
Фарфоровый тигель, доведенный до постоянной массы, ставят на глянцевую (лучше черную) бумагу. Осторожно извлекают из воронки высушенный фильтр с осадком и, держа над тиглем, свертывают. После этого аккуратно укладывают его в тигель. Если при внимательном осмотре на воронке обнаруживают следы осадка, то тщательно вытирают внутреннюю поверхность ее кусочком беззольного фильтра, который помещают в тот же тигель. Наконец, крупинки осадка, просыпавшиеся на бумагу при свертывании фильтра, также стряхивают в тигель. Затем ставят тигель на электрическую плитку и осторожно озоляют (сжигают) фильтр. Иногда вместо этого тигель вставляют в фарфоровый треугольник на кольце штатива и нагревают на небольшом пламени горелки. Желательно, чтобы фильтр медленно обуглился и истлел, не вспыхивая, так как горение приводит к потере мельчайших частиц осадка. Если он все-таки загорится, то пламя ни в коем случае не задувают, а только перестают нагревать и ждут, когда горение прекратится.
Закончив озоление фильтра, переносят тигель в муфельную печь и прокаливают 25—30 мин. Охлаждают тигель в эксикаторе, взвешивают и записывают значение его массы в лабораторный журнал. Повторяют прокаливание (15—20 мин), охлаждение и взвешивание до тех пор, пока не будет достигнута постоянная масса тигля с осадком.
2. Прокаливание осадка с отделением фильтра. К этому способу прибегают, когда осадок при обугливании фильтра может химически взаимодействовать с углеродом (восстанавливаться). Например, осадок хлорида серебра AgCl восстанавливается углеродом до свободного серебра; прокаливать его вместе с фильтром нельзя.
Хорошо высушенный осадок возможно полнее высыпают из фильтра на глянцевую бумагу и накрывают химическим стаканом (или опрокинутой воронкой), чтобы предотвратить потери. Фильтр с оставшимися на нем частицами осадка укладывают в тигель (доведенный до постоянной массы), сжигают и прокаливают. К прокаленному остатку в том же тигле присоединяют ранее отделенный осадок. После этого, как обычно, содержимое тигля прокаливают до постоянной массы.
Если осадок отфильтровывают с помощью стеклянного тигля, то вместо прокаливания прибегают к высушиванию до постоянной массы. Разумеется, фильтрующий тигель должен быть предварительно доведен до постоянной массы при той же температуре.
Если в ходе анализа будет допущена непоправимая ошибка (например, потеряна часть осадка, пролита часть раствора с осадком и т. п.), то определение следует начать снова, не расходуя время на получение заведомо неверного результата.
8. Взвешивание
Взвешивание производят на аналитических весах с точностью до 10-6 г. (ВЛР 200)
9. Вычисления в гравиметрическом анализе
Выше уже были рассмотрены некоторые сравнительно простые вычисления, а именно: расчет величины навески и количества осадителя, нахождение относительной ошибки определения. Вычисление результатов анализа также не отличается сложностью.
Обычно результаты гравиметрических определений выражают в процентах от исходного количества вещества. Для этого нужно знать величину навески анализируемого вещества, массу полученного осадка и его химическую формулу.
Гравиметрические определения преследуют различные цели. В одних случаях необходимо определить содержание того или иного элемента в химически чистом веществе, например содержание бария в хлориде бария BaCl2•2H2O. В других случаях требуется найти содержание действующего начала в каком-нибудь техническом продукте или вообще в веществе, имеющем примеси. Например, приходится определять содержание хлорида бария BaCl2•2H2O в продажном хлориде бария.
Техника определений в обоих приведенных случаях может оставаться одинаковой, но вычисления при этом будут различны. Рассмотрим ход вычислений на конкретных примерах.
Пример 1. Определить содержание чистого BaCl2•2H2O в образце технического хлорида бария. Навеска составляет 0,5956 г. Масса осадка сульфата бария BaSO4 после прокаливания равна 0,4646 г.
Решение: Определение основано на реакции, протекающей по уравнению
BaCl2•2H2O + H2SO4 = BaSO4 + 2HCl + 2H2O
М=244,30 г/моль М= 233,40 г/моль
Прежде всего вычисляют, какому количеству BaCl2•2H2O соответствует найденное в анализе количество BaSO4:
| 233,40 г | BaSO4 | получаются из | 244,30 г | BaCl2•2H2O |
| 0,4646 г | BaSO4 | получаются из | Х г | BaCl2•2H2O |
х = (0,4646 • 244.30)/233.40 = 0.4862 г BaCl2 • 2H2O.
Затем выражают содержание чистого BaCl2•2H2O в процентах от исходной навески технического хлорида бария:
| 0,5956 г | технического продукта | составляют | 100 % |
| 0,4862 г | чистого BaCl2•2H2O | составляют | х % |
х = (0,4862•100)/0.5956 = 81.83%.
Ответ: технический хлорид бария содержит 81.83% чистого BaCl2•2H2O.
Пример 2. Определить содержание бария в образце химически чистого хлорида бария BaCl2•2H2O. Навеска чистого BaCl2•2H2O равна 0,4872 г. Масса осадка сульфата бария BaSO4 после прокаливания 0.4644 г.
Решение. Сначала вычисляют, сколько бария (атомная масса 137,40) содержится в полученном осадке сульфата бария:
| в 233,40 г | BaSO4 | содержится | 137,40 г | Ba |
| в 0,4644 г | BaSO4 | содержится | х г | Ba |
х = (0,4644•137,40)/233,40 = 0,2733 г.
Очевидно, что это же количество бария входило до реакции в состав навески BaCl2•2H2O. Поэтому можно составить пропорцию:
| 0,4872 г | BaCl2•2H2O | составляют | 100 % |
| 0,2733 г | Ba | составляют | х % |
х = (0,2733 • 100)/0,4872 = 56,09%
Ответ: следовательно, хлорид бария BaCl2•2H2O содержит 56.09% бария.
Нередко для вычислений в гравиметрическом анализе используют факторы пересчета, называемые также аналитическими или весовыми факторами.
Фактор пересчета (F) представляет собой отношение атомной (или молекулярной) массы определяемого вещества к молекулярной массе вещества, находящегося в осадке:

Фактор пересчета показывает, сколько граммов определяемого вещества содержит 1 г осадка. В конкретных случаях факторы пересчета находят следующим образом:

При определении бария путем взвешивания в виде сульфата BaSO4 фактор пересчета равен:
F = ABa/MBaSO4 = 137,40 / 233,40 = 0,5887
Пользуясь факторами пересчета, делают вычисления по готовым формулам. Например, чтобы вычислить содержание элемента (или другой составной части) в сложном веществе, используют формулу:
% = (mF/G)•100,
где m— масса полученного осадка, г;
F — фактор пересчета;
G — навеска исследуемого вещества, г.
По этой формуле можно рассчитать и процентное содержание бария в хлориде бария BaCl2•2H2O:
Ba, % = (mF/C)•100 = [(0,4644•0,5887)/0,4872] • 100 = 56,09 %.
Суммирование погрешностей
В химических методах количественного химического анализа -гравиметрии и тириметрии в расчетах используют в основном суммы, разности, произведения и частные измеренных величин, определение каждой из которых содержит свою погрешность. Возникает задача вычисления суммарной погрешности, решение которой зависит от вида погрешностей и выполняемых арифметических действий с полученными значениями. В данном пособии рассмотрим суммирование погрешностей только для перечисленных арифметических действий. Правила суммирования представлены в табл. 9.1.
Таблица 9.1
Правила суммирования погрешностей
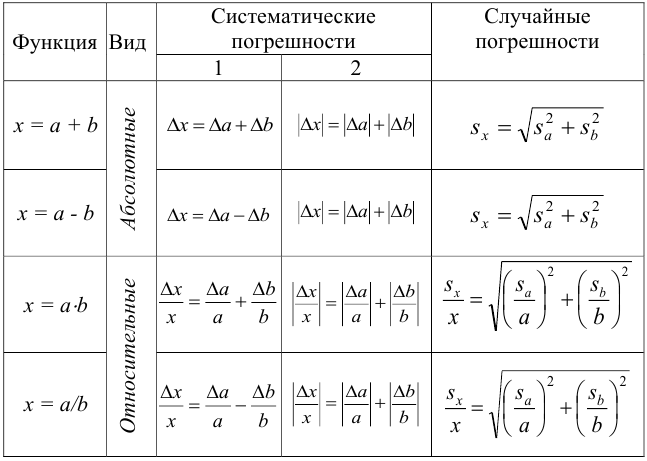
Примечание: суммарную систематическую погрешность рассчитывают по формулам столбца 1, если известны и величина, и знаки отдельных составляющих; если знаки неизвестны, расчет проводят по формулам столбца 2. Определив абсолютную погрешность, можно рассчитать относительную и наоборот.
Пример 9.8.
Вычислите абсолютную и относительную систематические погрешности взвешивания гравиметрической формы 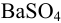 , если масса тигля с прокаленным осадком
, если масса тигля с прокаленным осадком 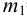 =12,3383 г, а пустого тигля
=12,3383 г, а пустого тигля 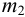 = 12,0112 г. Соответствующие систематические погрешности взвешивания с использованием аналитических разновесов составили
= 12,0112 г. Соответствующие систематические погрешности взвешивания с использованием аналитических разновесов составили 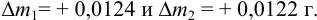
Решение:
Массу гравиметрической формы вычисляем по разности взвешиваний:
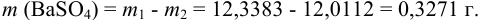
Используя правило сложения систематических погрешностей для разности при известном знаке составляющих (колонка 1 табл. 9.1), получаем:
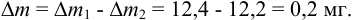
Следует обратить внимание на то, что при использовании одних и тех же гирь при взвешивании тигля с осадком и без него систематическая погрешность уменьшается.
Найдем относительную погрешность определения массы гравиметрической формы:
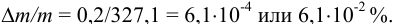
Пример 9.9.
Рассчитайте систематическую погрешность (абсолютную и относительную) концентрации 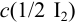 при приготовлении 250,0 мл раствора иода из навески /и 1,2634 г. Систематическая погрешность массы навески +0,4 мг, измерения объема мерной колбой -0,2 мл.
при приготовлении 250,0 мл раствора иода из навески /и 1,2634 г. Систематическая погрешность массы навески +0,4 мг, измерения объема мерной колбой -0,2 мл.
Решение:
Концентрацию раствора рассчитываем по формуле:
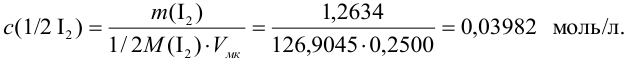
В соответствии с законом распространения систематических погрешностей относительная погрешность частного при известных знаках определяется равна разности относительных погрешностей делимого и делителя, а произведения — сумме (табл. 9.1, колонка 1):
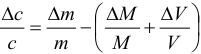
Поскольку погрешность определения молярной массы мала по сравнению с другими погрешностями, ее вкладом пренебрегаем. Найдем относительную погрешность величины концентрации:
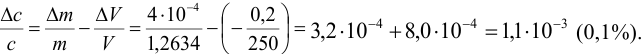
Абсолютная погрешность концентрации составляет:
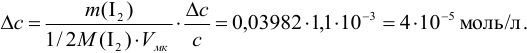
Пример 9.10.
Рассчитайте максимально допустимое относительное стандартное отклонение при определении массовой доли серы в пересчете на 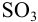 весовым методом, если методика предполагает: из навески пробы около 1 г после разложения и окисления серы до
весовым методом, если методика предполагает: из навески пробы около 1 г после разложения и окисления серы до 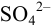 остаток перевести в мерную колбу вместимостью 100 мл, осаждение провести из аликвоты 20 мл и получить массу гравиметрической формы 0,4 г (см. гл. 7). Использована посуда второго класса (табл. 11 приложения).
остаток перевести в мерную колбу вместимостью 100 мл, осаждение провести из аликвоты 20 мл и получить массу гравиметрической формы 0,4 г (см. гл. 7). Использована посуда второго класса (табл. 11 приложения).
Решение:
При осаждении из аликвоты результаты весового анализа могут быть рассчитаны по формуле:
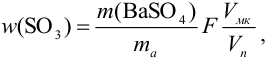
где 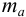 — масса пробы,
— масса пробы, 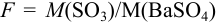 — гравиметрический фактор.
— гравиметрический фактор.
С учетом правила суммирования случайных погрешностей для произведения и частного (табл. 9.1) относительное стандартное отклонение результата можно выразить формулой:
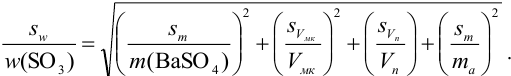
При этом пренебрегаем погрешностью гравиметрического фактора 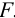
Масса гравиметрической формы 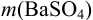 получается как разность двух взвешиваний на аналитических весах:
получается как разность двух взвешиваний на аналитических весах:
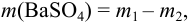
где — суммарная масса тигля и ; — масса пустого тигля.
Аналогично берут навеску пробы. Погрешность взвешивания примерно одинакова в обоих случаях: 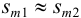 . Вычислим абсолютную случайную погрешность определения массы
. Вычислим абсолютную случайную погрешность определения массы 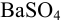 как погрешность разности:
как погрешность разности:
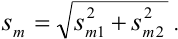
При взвешивании тигля до постоянной массы погрешность взвешивания составляет 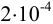 г. Тогда абсолютная погрешность определения массы:
г. Тогда абсолютная погрешность определения массы:
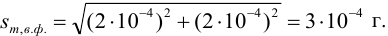
Абсолютная погрешность взятия навески иода на аналитических весах:
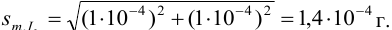
Из табл. 11 приложения берем соответствующие допуски мерной посуды и подставляя в формулу (9,6) вычисляем максимально возможное относительное стандартное отклонение результата:
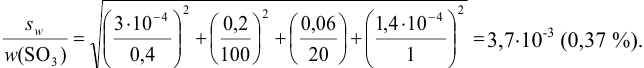
Пример 9.11.
Рассчитайте максимальную допустимую погрешность (абсолютную и относительную) концентрации 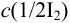 при приготовлении 250 мл раствора иода из навески
при приготовлении 250 мл раствора иода из навески 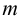 1,2634 г.
1,2634 г.
Решение:
Вычисление проводим с учетом примера 9.9, где найдена концентрация 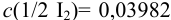 моль/л, используя формулу, подобную (9.6) и рассуждения примера 9.10. Основной вклад в суммарную погрешность концентрации иода вносят погрешность измерения объема и массы:
моль/л, используя формулу, подобную (9.6) и рассуждения примера 9.10. Основной вклад в суммарную погрешность концентрации иода вносят погрешность измерения объема и массы:

Очевидно, что измерение объема вносит больший вклад в погрешность. Вычислим максимально допустимую абсолютную погрешность:
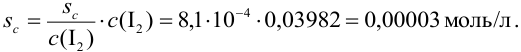
Пример 9.12.
Определите относительную и абсолютную погрешность концентрации раствора тиосульфата, если она устанавливалась по раствору иода, приготовленному как в примере 9.11, по результатам одного титрования аликвоты иода тиосульфатом (расход тиосульфата 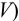 и трех параллельных титрований. Соответствующие данные (с указанием в скобках стандартных отклонений):
и трех параллельных титрований. Соответствующие данные (с указанием в скобках стандартных отклонений):

Решение:
Концентрацию тиосульфата вычисляем по формуле (гл. 8):
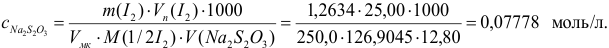
Расчет относительной погрешности проводим с учетом вклада всех измеряемых составляющих, упуская малую погрешность молярной массы:

Из расчета следует, что наибольший вклад в погрешность вносит объем титранта, и с его увеличением погрешность уменьшается. Если титрование повторять 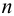 раз с повой аликвотой относительная погрешность концентрации уменьшится в
раз с повой аликвотой относительная погрешность концентрации уменьшится в 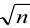 раз, т. е. при трех параллельных титрованиях:
раз, т. е. при трех параллельных титрованиях:
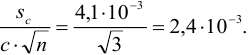
Абсолютная случайная погрешность определения концентрации  при однократном титровании:
при однократном титровании:
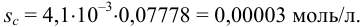
Пример 9.13.
Проведите расчет погрешности результата анализа образца на содержание 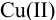 , проведенного йодометрическим титрованием по заместителю согласно реакциям:
, проведенного йодометрическим титрованием по заместителю согласно реакциям:
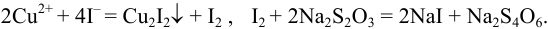
Навеску образца 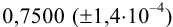 г перевели в мерную колбу вместимостью 200,0 (±0,2/3) мл. На титрование брали три аликвоты по 20,00 (±0,06/3) мл, расход тиосульфата с концентрацией 0,07778
г перевели в мерную колбу вместимостью 200,0 (±0,2/3) мл. На титрование брали три аликвоты по 20,00 (±0,06/3) мл, расход тиосульфата с концентрацией 0,07778 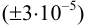 моль/л составил: 10,00; 10,05; 10,07; 10,03 (
моль/л составил: 10,00; 10,05; 10,07; 10,03 (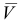 =10,04) мл.
=10,04) мл.
Решение:
Рассчитаем стандартное отклонение объема тиосульфата:
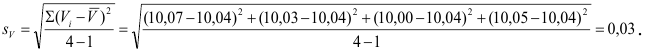
Массовую долю меди вычислим по формуле:
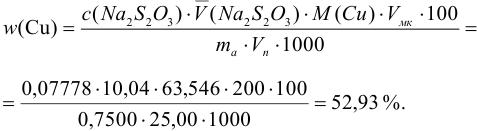
С учетом погрешностей входящих в формулу величин рассчитаем относительную погрешность содержания меди:
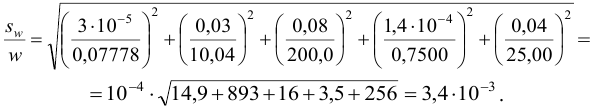
Абсолютная погрешность: 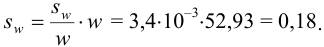
Тогда можно представить результат определения массовой доли меди как 53,27±0,18 (%).
Эти примеры взяты со страницы примеров решения задач по аналитической химии:
Решение задач по аналитической химии
Возможны вам будут полезны эти страницы: